#mitochondrial therapy
Explore tagged Tumblr posts
Visit Tumblr Blog
Explore Tumblr blogs with no restrictions, modern design and the best experience.
Last Seen Tumblr Blogs





Fun Fact
Tumblr was the first site to host the blog for President Barack Obama in 2011.
Text
youtube
#Spinal cord injury#neuroregeneration#nanomedicine#mitochondrial therapy#silica nanoparticles#dual-responsive nanoparticles#targeted drug delivery#neuroprotection#regenerative medicine#oxidative stress#inflammation control#apoptosis inhibition#cellular repair#tissue regeneration#advanced therapeutics#biomaterials#neuroscience research#precision medicine#theranostics#medical breakthrough.#Youtube
2 notes
·
View notes
Text
Mitochondrial Dysfunction in Spinal Muscular Atrophy (SMA)
Introduction
Spinal Muscular Atrophy (SMA) is a severe neurodegenerative disorder that predominantly affects motor neurons, resulting in progressive muscle weakness and atrophy. The condition is caused by mutations in the survival motor neuron 1 (SMN1) gene, which leads to the loss of SMN protein, a critical factor for motor neuron survival. Although the primary defect lies in the motor neurons, increasing evidence suggests that mitochondrial dysfunction plays a pivotal role in the pathophysiology of SMA. Mitochondria, the powerhouse of the cell, are crucial for cellular energy production and regulation of various metabolic pathways. In the context of SMA, mitochondrial dysfunction has been linked to impaired cellular energy metabolism, oxidative stress, and neuronal death.
This article reviews the emerging role of mitochondrial dysfunction in SMA, examining its impact on motor neurons, the cellular processes involved, and the potential for mitochondrial-targeted therapies.
Mitochondrial Dysfunction in SMA: A Pathophysiological Overview
Mitochondria are essential organelles responsible for generating ATP through oxidative phosphorylation, controlling cellular metabolism, and mediating cell death mechanisms. In SMA, deficits in SMN protein affect multiple cellular pathways, including mitochondrial function. SMN is known to be involved in the biogenesis and maintenance of mitochondria. When its expression is reduced, mitochondrial dysfunction occurs in several ways, contributing to the progressive nature of SMA.
Impaired Mitochondrial Biogenesis
Mitochondrial biogenesis refers to the process by which new mitochondria are formed within cells. This process is tightly regulated by nuclear and mitochondrial signals, with the peroxisome proliferator-activated receptor-gamma coactivator 1-alpha (PGC-1α) being a key regulator of mitochondrial biogenesis. Studies in SMA models have shown that a reduction in SMN protein leads to downregulation of PGC-1α, resulting in decreased mitochondrial biogenesis. This reduced mitochondrial mass is particularly detrimental to motor neurons, which have high energy demands due to their long axonal projections and rapid neurotransmitter signaling.
Mitochondrial Dysfunction and ATP Production
Mitochondrial dysfunction in SMA results in decreased ATP production. ATP is required for essential cellular functions such as protein synthesis, ion transport, and maintaining membrane potential. In motor neurons, impaired ATP generation leads to cellular energy deficits that exacerbate neurodegeneration. Mitochondrial dysfunction also disrupts calcium homeostasis, as mitochondria play a central role in buffering intracellular calcium levels. Elevated intracellular calcium levels can activate enzymes that degrade cellular components, further contributing to cell death in motor neurons.
Oxidative Stress
One of the most significant consequences of mitochondrial dysfunction is the increased production of reactive oxygen species (ROS). Mitochondria are the main source of ROS in cells, and under normal conditions, the antioxidant defense systems neutralize these reactive molecules. However, in SMA, defective mitochondrial function leads to excessive ROS production, which overwhelms the cell’s ability to detoxify them. ROS are highly reactive and can damage cellular structures such as proteins, lipids, and DNA, ultimately contributing to oxidative stress and neuronal injury.
Mitochondrial Dynamics and Morphology
Mitochondrial morphology is highly dynamic, with the organelles undergoing fusion and fission events in response to cellular needs. In SMA, the balance between these processes is disrupted. Studies have shown that reduced SMN levels lead to an increase in mitochondrial fragmentation, a characteristic of mitochondrial dysfunction. Fragmented mitochondria are less efficient in energy production and more prone to damage. Additionally, the fragmented mitochondria in SMA models exhibit impaired mitochondrial transport along axons, further hindering motor neuron function.
Mitochondrial Quality Control
Mitochondrial quality control mechanisms, such as mitophagy, are critical for maintaining mitochondrial health. Mitophagy is the process by which damaged mitochondria are selectively degraded by autophagosomes. In SMA, defects in SMN protein affect the cellular machinery responsible for mitophagy, leading to the accumulation of dysfunctional mitochondria. This impairment in mitochondrial turnover accelerates neurodegeneration by allowing damaged mitochondria to persist, increasing oxidative stress, and triggering cellular apoptosis.
Mitochondrial Dysfunction in Different Types of SMA
SMA is classified into several types based on age of onset and severity, including Type I (Werdnig-Hoffmann disease), Type II, Type III, and Type IV. Mitochondrial dysfunction is observed in all types, but its extent varies depending on the severity of the disease.
SMA Type I
This is the most severe form of SMA, typically presenting in infants before six months of age. These children experience profound muscle weakness and may not survive beyond the first two years of life without intervention. In Type I, mitochondrial dysfunction is particularly pronounced, with severe mitochondrial fragmentation, impaired ATP production, and significant oxidative damage observed in motor neurons. The severity of mitochondrial dysfunction correlates with the extent of neurodegeneration in the spinal cord.
SMA Type II
Type II SMA presents later in infancy or early childhood, with affected individuals showing progressive muscle weakness but with a longer life expectancy compared to Type I. Mitochondrial dysfunction in Type II is still significant but less severe than in Type I. There is evidence of mitochondrial fragmentation and altered mitochondrial dynamics, but motor neurons in Type II patients may still retain some capacity for mitochondrial biogenesis and ATP production, contributing to the slower progression of the disease.
SMA Type III and IV
SMA Type III and IV are milder forms of the disease, with onset typically in childhood or adulthood. While mitochondrial dysfunction is present, it is less pronounced than in Type I and II. In these types, mitochondrial dynamics, ATP production, and oxidative stress are affected, but the clinical presentation is less severe, and individuals often experience a normal or near-normal life expectancy.
Conclusion
Mitochondrial dysfunction is a central feature of the pathophysiology of Spinal Muscular Atrophy (SMA). Reduced SMN protein leads to impaired mitochondrial biogenesis, altered mitochondrial dynamics, increased oxidative stress, and mitochondrial dysfunction. These defects contribute to the progressive degeneration of motor neurons and muscle weakness seen in SMA. Understanding the complex interplay between SMN deficiency and mitochondrial dysfunction provides valuable insights into the disease mechanisms and offers new avenues for therapeutic intervention. Mitochondrial-targeted approaches, including enhancing mitochondrial biogenesis, antioxidant therapy, and modulation of mitochondrial dynamics, hold promise for improving the quality of life and outcomes for SMA patients.
Ongoing research into mitochondrial dysfunction in SMA is crucial for identifying novel treatment strategies that can complement existing therapies and slow disease progression. As therapeutic options evolve, mitochondrial health will likely become an important consideration in the management of SMA, offering hope for more effective treatments in the future.

#Spinal Muscular Atrophy (SMA)#Mitochondrial Dysfunction#Motor Neurons#SMN1 Gene#SMN Protein#Neurodegeneration#Mitochondrial Biogenesis#Oxidative Stress#ATP Production#Mitochondrial Fragmentation#Reactive Oxygen Species (ROS)#Calcium Homeostasis#Mitochondrial Dynamics#Mitochondrial Transport#Mitophagy#Mitochondrial Quality Control#PGC-1α (Peroxisome Proliferator-Activated Receptor-Gamma Coactivator 1-Alpha)#Cellular Energy Metabolism#Mitochondrial-Targeted Therapies#Apoptosis.
0 notes
Text
Also preserved on our archive
Some interesting science analyzed
BY BROOKS LEITNER
Imagine lying back in an enclosed chamber where you bask for 90 minutes in a sea of pure oxygen at pressures two to three times that felt at sea level. This is the world of hyperbaric oxygen therapy (HBOT), a technology that’s been around for decades and is now being explored as a possible treatment for Long COVID.
"The silence on the inside is deafening at first,” says John M.,* who has undergone dozens of HBOT treatments for his persistent Long COVID symptoms. Fortunately, there is a television outside the chamber in view, and it is easy to communicate with the provider if needed. While the potential protocol is still being refined, patients may undergo up to 40 HBOT sessions to address some of the most problematic, lingering symptoms of this complex condition.
HBOT is a therapeutic process that has been widely used to treat such conditions as decompression sickness in scuba divers, carbon monoxide poisoning, and diabetic foot ulcers. In HBOT, the body is exposed to 100% oxygen, a significant increase from the 21% oxygen concentration we typically breathe. The therapy takes place in an enclosed chamber where the air pressure is elevated above normal levels. The combination of high-pressure and high-oxygen conditions enhances the amount of oxygen that can reach the body's tissues. The hope is that this therapy can provide the same relief and healing to people with Long COVID that it does for those with other conditions.
According to John M., HBOT was the first treatment that helped with his sleep and reduced his heart palpitations. “At one point after hospitalization, my Long COVID symptoms were so bad that I could barely walk or talk. HBOT was a great tool that really assisted with my recovery,” he said. John added that he hopes the medical community will achieve a better understanding of how HBOT can help relieve suffering for patients with Long COVID and that more research will increase access to this innovative therapy.
Does HBOT improve Long COVID symptoms? One key observation from the work of Inderjit Singh, MBChB, an assistant professor at Yale School of Medicine (YSM) specializing in pulmonary, critical care, and sleep medicine, is that Long COVID patients often experience debilitating fatigue. Based on Dr. Singh’s previous Long COVID research, the exhaustion is thought to be linked to the muscles’ inability to efficiently extract and utilize oxygen.
To picture how HBOT might work, you can think of your muscles as engines sputtering, struggling to get the fuel they need. If oxygen is the gas that fuels the muscles, it’s as if you are trying to complete your daily routine while the gas tank is running on “empty.” By aiming to directly address this oxygen utilization impairment, HBOT may be a potential solution.
A systematic review by researchers at the China Medical University Hospital noted that HBOT could tackle another major factor in the Long COVID puzzle: oxidative stress. This relates to the body's struggle to maintain balance when harmful molecules, known as free radicals, run amok, causing chronic inflammation.
Research co-authored by Sandra K. Wainwright, MD, medical director of the Center for Hyperbaric Medicine and Wound Healing at Greenwich Hospital in Connecticut, suggests that HBOT, with its high-oxygen environment, might dampen this chronic inflammation by improving mitochondrial activity and decreasing production of harmful molecules. Other potential benefits of HBOT in the treatment of Long COVID may include restoration of oxygen to oxygen-starved tissues, reduced production of inflammatory cytokines, and increased mobilization of hematopoietic stem cells—primary cells that transform into red blood cells, white blood cells, and platelets.
HBOT for Long COVID: Current and ongoing research Several small-scale reports have indicated that HBOT is safe for patients with Long COVID.
To address this question, a trial that followed the gold standard of modern medical research—a randomized, placebo-controlled, double-blind design—assigned 73 Long COVID patients to either receive 40 sessions of HBOT or a placebo of only 21% oxygen. The study observed positive changes in attention, sleep quality, pain symptoms, and energy levels among participants receiving HBOT. In a longitudinal follow-up study published in Scientific Reports, the authors at the Tel Aviv University found that clinical improvements persisted even one year after the last HBOT session was concluded. In a second study, the same authors focused on heart function, measured by an echocardiogram, and found a significant reduction in heart strain, known as global longitudinal strain, in patients who received HBOT.
In another study, 10 patients with Long COVID underwent 10 HBOT treatments over 12 consecutive days. Testing showed statistically significant improvement in fatigue and cognitive function. Meanwhile, an ongoing trial at the Karolinska Institute in Sweden has reported interim safety results wherein almost half of the Long COVID patients in the trial reported cough or chest discomfort during treatment. However, it was unclear whether HBOT exacerbated this symptom or if this adverse effect was due to the effort of participation by patients suffering from more severe Long COVID symptoms.
Is HBOT currently available as a treatment for Long COVID? For HBOT to become a mainstream treatment option for Long COVID, several critical priorities must be addressed. First, there is currently no established method for tailoring HBOT dosages to individual patients, so researchers must learn more about the specific features or symptoms that indicate potential benefits from HBOT. At the same time, we need to identify factors that may be associated with any adverse outcomes of HBOT. And finally, it’s important to determine how long these potentially beneficial effects last in a larger cohort. Will just a few HBOT trials be enough to restore patients to their baseline health, or will HBOT become a recurring component of their annual treatment regimen?
For now, HBOT remains an experimental therapy—and as such is not covered by insurance. This is a huge issue for patients because the therapy is expensive. According to Dr. Wainwright, a six-week course of therapy can run around $60,000. That’s a lot to pay for a therapy that’s still being studied. In the current completed studies, different treatment frequencies and intensities have been used, but it’s unclear how the treatment conditions affect the patient’s outcome.
“I have had some patients notice improvements after only 10 or 15 treatments, whereas some others need up to 45 treatments before they notice a difference,” notes Dr. Wainwright. “I think that HBOT is offering some promising results in many patients, but it is probably a strong adjunctive treatment to the other spectrum of things Long COVID patients should be doing, like participating in an exercise, rehab, and nutritional program.”
Dr. Singh notes that “a major challenge for research is the heterogeneity of Long COVID. It is hard to determine which symptoms to treat and enroll patients into trials based on them.”
Perhaps treatments that target multiple issues caused by Long COVID, like HBOT, may help overcome this challenge.
*Not his real name.
Brooks Leitner is an MD/PhD candidate at Yale School of Medicine.
The last word from Lisa Sanders, MD: Hyperbaric oxygen therapy (HBOT) is just one of the many existing treatments that are being looked at to treat Long COVID. We see this with many new diseases—trying to use a treatment that is effective in one set of diseases to treat another. And there is reason for optimism: We know that HBOT can deliver high levels of oxygen to tissues in need of oxygen. That’s why it’s used to treat soft tissue wounds. If reduced oxygen uptake is the cause of the devastating fatigue caused by Long COVID, as is suggested by many studies, then perhaps a better delivery system will help at least some patients.
Studies referenced:
bmcinfectdis.biomedcentral.com/articles/10.1186/s12879-023-08002-8
www.ncbi.nlm.nih.gov/pmc/articles/PMC8806311/
www.nature.com/articles/s41598-024-53091-3
www.nature.com/articles/s41598-022-15565-0
www.frontiersin.org/journals/medicine/articles/10.3389/fmed.2024.1354088/full
www.ncbi.nlm.nih.gov/pmc/articles/PMC11051078/#:~:text=Proposed%20Mechanism%20of%20HBOT%20o
#long covid#hbottherapy#HBOT#hyperbaric oxygen therapy#mask up#covid#pandemic#wear a mask#public health#covid 19#still coviding#wear a respirator#coronavirus#sars cov 2
37 notes
·
View notes
Note
I hope you don’t mind me asking but did your doctors tell you when they suspected you had a life-limiting diagnosis? And if so how did you keep yourself from going crazy during that in between time?
My doctors are concerned about neuromuscular disorders and I’m currently driving myself up a wall.
They thought I was crazy literally up until the results of my muscle biopsy, unfortunately. And I was going crazy, just not in the way they thought. I was obsessed with the idea that I was dying and would die before I got a diagnosis.
One thing that helps me keep from stressing now is looking up statistics and how things affect life length and quality. Make sure you find out whether or not they're including ALS in those statistics or if it's for other neuromuscular diseases, the ALS statistics shorten the life expectancy from diagnosis data by a LOT compared to other neuromuscular diseases. I'm a lot less scared when I see 12 years average from needing a trach rather than 4 years, for example. Another thing to consider is that you're probably done growing, and if you end up having mitochondrial disease or muscular dystrophy that GREATLY increases life expectancy.
The thing that's most likely gonna kill me is respiratory failure and the biggest way to prevent an earlier death than necessary is to be willing and compliant when it comes to using things like a ventilator.
Get a lung function test, get an echocardiogram, and get a swallow study. Make sure that you have the adaptive equipment you need to avoid pressure sores. These are the biggest issues that affect neuromuscular patients and avoiding them can keep you alive for a lot longer.
Finally, find what's important in life *to you* and work on making sure you can do it as long as possible. For me, that's my garden, so when I do physical therapy, I work on keeping the skills I need for that verses trying to build new skills that I'm going to lose shortly after anyways. I also work on finding tools that I can use to make it easier, those aren't always adaptive equipment specifically. I'm getting a stand up weeder tool soon that I use from my wheelchair without bending over. Gardening is really nice because it's something *alive* and it's alive because of my work and a tangible difference in the world that I made. I've planted trees that will outlive me and that's really comforting. It doesn't have to be gardening though, I have severely disabled friends who keep snail tanks and guinea pigs, and it has the same sorts of benefits.
It's scary, it sucks, I STILL stress out a lot about it at times. But what matters is living a life I find worth living with the time that I have and that helps me find peace. I hope you find that peace too.
32 notes
·
View notes
Text
things that people who are stop recommending to CFS/ME/SEID/long COVID with PEM and CF (called "PEM w/ CF" henceforth) patients immediately:
Exercise. This includes yoga, pilates, aquatherapy, and other "gentle" exercises. If it works against gravity or another resistance, it's not good for treating PEM w/ CF. The deconditioning theory was debunked years ago, so don't bring that shit here.
B-12. It's important to get tested for a B-12 deficiency as the symptoms can be similar to PEM w/ CF, but once it's ruled out added B-12 can have side effects. The main and most studied one is insomnia, but anecdotal reports (including my own) also often mention racing heart rate and jitters. Since these side effects use up energy, it's better for people with PEM w/ CF to err on the side of caution and avoid excess B-12 intake. Also it's pretty much the first thing anyone suggests so unless someone is newly ill they've likely already tried it and are sick of hearing about it.
Caffeine and other stimulants. While these can make you feel energized temporarily, it doesn't actually increase the amount of energy in your body. Assuming we're running on the theory of PEM w/ CF being a deficiency of ATP caused by mitochondrial issues, fatigue isn't a state of mind. It's our bodies telling us we're running out of fuel to keep them going. Also many of us have a co-morbid tachycardia condition and a flair up of that condition can cause a flair up of fatigue.
Medication and other treatments designed for mental illnesses. While having a chronic illness can cause mental illness, especially if you live in an ableist country with failed disability aid services, mental illness does not cause PEM w/ CF. Treatment for mental illness won't actually treat the symptoms of PEM w/ CF.
Alternative medicine. Many alternative medicine treatments are actively dangerous and the idea that there's "no harm in trying" because "it's natural anyway" is false. Stop recommending ozone therapy (potentially deadly and not useful in concentrations not strong enough to kill you), stop recommended chiropractors (can result in a stroke), stop recommending essential oils (no approved medical use beyond basic relaxation and concentrated nature can make them dangerous), you get the picture. Alternative medicine preys on desperate suffering people to sell them ineffective, potentially dangerous "treatments", don't do their advertising for them.
PEM w/ CF patients feel free to add on treatments that are commonly recommended but unscientific/uneffective, others be respectful and don't derail.
#cfs#chronic fatigue syndrome#me/cfs#cfs/me#seid#long covid#cpunk#cripplepunk#cripple punk#actually disabled
94 notes
·
View notes
Text
Moon Missions
What’s going on with the moon?
The United States recently had a solar eclipse on April 8th, 2024, and some might be surprised to learn that the moon is, in fact, affected by solar radiation. The charged particles emitted by the sun, called the solar wind, reach the moon with no interruption from its atmosphere, as it has none. It also has no global magnetic field, another layer of protection that Earth does have, in comparison.
The moon does, however, have small areas of magnetic fields. We can see this because these areas remain lighter in photos whereas chemical reactions from radiation darken the unprotected areas.
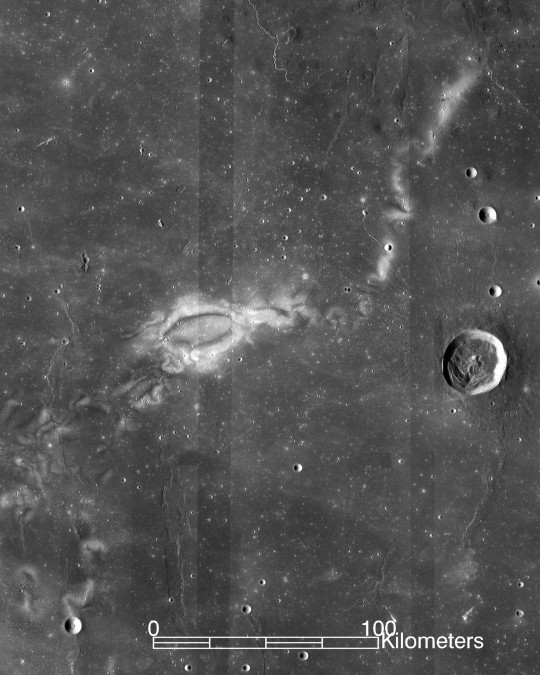
Fortunately, most of these charged particles cannot pass through the hulls of space stations, so astronauts are safe in orbit. Cosmic rays, made of stronger and faster-moving particles, are more dangerous. Even on Earth, under the atmosphere and magnetosphere, cosmic radiation reaches humans, though not enough to be considered damaging to our health.
A lander and rover launched in 2018 delivered the first measurements of radiation levels on the moon 4. Based on those data, astronauts on the moon can be exposed to up to 150 times higher radiation levels than on Earth.
Radiation is a leading reason for the pause in lunar landing missions. It raises risks of cataracts, heart diseases, radiation illness, cancer, and other ailments. Longer missions, of course, would heavily exacerbate these radiation doses.
Other Health Concerns
Cosmic rays contain High-Energy (HZE) ions. In different exposure such as from nuclear accidents or irradiation therapy, HZE ions have been found to cause dysregulation in the mitochondria and damage to DNA. Because of this, prolonged exposure is linked to health effects often associated with aging, such as hippocampus synapse loss and metabolic disruption caused by damage to mitochondrial DNA.
Long-duration space flights have also been linked to cardiovascular disorders. For astronauts on the Apollo missions, heart attack was “the second leading cause of death” 8. For additional space flights outside of Earth’s magnetosphere, astronauts also had a higher mortality rate due to cardiovascular diseases.
In a previous article, we discussed the relationship between circadian rhythms and health. These rhythms are another thing that space travel can impact, causing sleep and mental health disturbances in astronauts 9.
While various studies are investigating the conditions of these health risks, a current NASA mission is specifically investigating radiation protection.
Long-term Mission
NASA plans on eventually returning to human-manned missions to the moon.
First, they have to address the issues discussed above.
In November of 2022, Artemis I launched with two manikins bearing radiation detectors. From this mission, NASA was able to confirm the success of the intended trajectory, launch of ground systems, and the Orion spacecraft. The radiation results from this mission are still being analyzed.
The Artemis missions are intended to explore more of the moon than ever before, and lay groundwork for eventual missions to Mars.
Artemis II will not launch any earlier than September of 2025. It is planned to last ten days, consist of a 4-person crew, and be a lunar flyby to ensure the proper functioning of the spacecraft’s systems.
It has seemed for years that lunar exploration has halted. Manned missions have indeed been paused, for good reasons. Ensuring the safety of astronauts is a priority, and they face serious health risks even when missions go as expected. But NASA intends to continue exploring space, the moon, and Mars. The current Artemis missions are discovering improved, new ways to ensure the safety of astronauts while making scientific progress.
Additional Resources
1. https://science.nasa.gov/moon/solar-wind/
2. https://phys.org/news/2012-01-solar-flares-astronauts.html
3. https://arxiv.org/ftp/arxiv/papers/1211/1211.3962.pdf
4. https://link.springer.com/article/10.1007/s11214-020-00725-3
5.https://www.nasa.gov/missions/artemis/orion/orion-passengers-on-artemis-i-to-test-radiation-vest-for-deep-space-missions/
6.https://www.smithsonianmag.com/science-nature/how-space-radiation-threatens-lunar-exploration-180981415/
7.https://www.nasa.gov/humans-in-space/analysis-confirms-successful-artemis-i-moon-mission-reviews-continue-2/
8.https://www.frontiersin.org/journals/physiology/articles/10.3389/fphys.2020.00955/full
9. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9818606/
#radiation#moon#moon mission#nasa#nasa photos#article#research#solar wind#solar radiation#cosmic rays#cosmic radiation#space exploration#science#space#Work has been horribly busy so the next article might not be out on the usual schedule
19 notes
·
View notes
Text
i've been collecting resources and studies about myalgic encephalomyelitis for upcoming doctors appointments, and since there's a lot of misinformation about ME out there i thought i'd make a post with links and information that might be helpful for other ME patients, or just generally educational!
for anyone who doesn't know, myalgic encephalomyelitis is a debilitating multi system chronic illness. it's most characteristic symptom is post exertional malaise/symptom exacerbation (meaning symptoms getting significantly worse after exertion with prolonged recovery periods - see the diagnostic criteria linked below for more explanation). it has previously been called chronic fatigue syndrome, a name associated with claims of it being a psychosomatic condition rather than a medical one despite evidence to the contrary (here's a page with some information about the history of ME, and see the pathology section further down for evidence of ME being a physical disease). ME is also often comorbid with conditions like POTS (or other forms of orthostatic intolerance and dysautonomia), MCAS, and Small Fibre Neuropathy, and it's not uncommon for people who have long covid to develop ME. but yea! here's some links to resources i've gathered.
general resources/overviews:
Chronic Fatigue Syndrome Myalgic Encephalomyelitis Primer For Clinical Practitioners 2014 Edition
Diagnosis and Management of Myalgic Encephalomyelitis - ME Action
Initiating Care of a Patient With Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS)
Medical considerations when treating urgently ill patients with underlying myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS)
Caring for the Patient with Severe or Very Severe Myalgic Encephalomyelitis/Chronic Fatigue Syndrome
Three Cases of Severe ME/CFS in Adults
diagnostics:
Myalgic Encephalomyelitis: International Consensus Criteria <- just the criteria
Myalgic encephalomyelitis: International Consensus Criteria <- criteria with explanation of how and why it was developed
TESTING RECOMMENDATIONS FOR SUSPECTED ME/CFS US ME/CFS Clinician Coalition
there are multiple ME severity scales, and exact definitions of what constitutes mild/moderate/severe/very severe vary a bit, but here is one: M.E. Disability Scale, another one is the ME/CFS Disability Rating Scale from ME Association, however the pdf on their website costs so i also have a pdf that i made with the text
pathology:
Brainstem volume changes in myalgic encephalomyelitis/chronic fatigue syndrome and long COVID patients
Decreased oxygen extraction during cardiopulmonary exercise test in patients with chronic fatigue syndrome
Developing a blood cell-based diagnostic test for myalgic encephalomyelitis/chronic fatigue syndrome using peripheral blood mononuclear cells
Human Herpesvirus-6 Reactivation, Mitochondrial Fragmentation, and the Coordination of Antiviral and Metabolic Phenotypes in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome
Mitochondrial complex activity in permeabilised cells of chronic fatigue syndrome patients using two cell types
Muscle sodium content in patients with Myalgic Encephalomyelitis/Chronic Fatigue Syndrome
Redox imbalance links COVID-19 and myalgic encephalomyelitis/chronic fatigue syndrome
The use of oxygen as a possible screening biomarker for the diagnosis of chronic fatigue
Tissue specific signature of HHV-6 infection in ME/CFS
treatment (both helpful and harmful):
ME/CFS TREATMENT RECOMMENDATIONS US ME/CFS Clinician Coalition
Low-dose naltrexone in the treatment of myalgic encephalomyelitis/chronic fatigue syndrome (ME/ CFS)
Potential Therapeutic Benefit of Low Dose Naltrexone in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: Role of Transient Receptor Potential Melastatin 3 Ion Channels in Pathophysiology and Treatment
Back to the Future? Immunoglobulin Therapy for Myalgic Encephalomyelitis/Chronic Fatigue Syndrome
Evidence Against Exercise for people with PEM/PESE in Long COVID and ME/CFS
PACE trial claims for recovery in myalgic encephalomyelitis/chronic fatigue syndrome – true or false? It’s time for an independent review of the methodology and results
Treatment harms to patients with ME/CFS
#more than okay to reblog!#🐛#myalgic encephalomyelitis#me/cfs#chronic fatigue syndrome#chronic illness#disability#actually disabled#long covid
143 notes
·
View notes
Text
This place make my mind and body happy!!! @upgradelabsriverton 😁 The therapies here are designed to tame inflammation and promote healing on a cellular level. This is beneficial for anyone with inflammation, or athletes needing recovery. I often point my clients to these therapies because of their science-backed benefits. Here are some I enjoyed today....
RED LIGHT THERAPY - improves cellular function and health by enhancing mitochondrial function and improving the production of ATP (cells energy source)
LYMPHATIC MASSAGE (the big squeeze) - stimulates the lymphatic system responsible for transporting white blood cells to help fight infection and strengthen immune system
CRYOTHERAPY (cold therapy) - lowers oxidative stress (free radicals) and inflammation by reducing the production and release of pro-inflammatory substances (eg. cytokines) while increasing the release of anti-inflammatory substances (noradrenaline)
If this Hawaiian can do cryotherapy, so can you! Thank you Tom, Prosper & Nate 🤙🏽
#biohacking#cellhealth#inflammation#antiinflammatory#health#Healthy#longevity#healthyliving#healthylifestyle#recovery#cognitive#brain health
2 notes
·
View notes
Text
Dr. Thomas Seyfried: Cancer as a Mitochondrial Metabolic Disease
youtube
1 - Cancer is a type of metabolic disease: It is not a genetic disease!
2 - A reliance on substrate level phosphorylation for energy is the metabolic hallmark of all or nearly all cancers.
3 - The simultaneous restriction of glucose and glutamine can manage GBM and most other cancers.
4 - Press-Pulse metabolic therapy is a non-toxic, cost effective strategy for the management and possible resolution of all types of cancer!
Cancer can be addressed with a combination of calorie restriction and keto diet.
5 notes
·
View notes
Text
Meet the Amatruda Lab!
James Amatruda, MD, PhD

www.chla.org
Dr. James Amatruda is the Head of Basic and Translational Research for the Cancer and Blood Disease Institute at Children’s Hospital Los Angeles. He’s the inaugural holder of the Dr. Kenneth O. Williams Chair in Cancer Research. Dr. Amatruda is a Professor of Pediatrics and Medicine for the Keck School of Medicine of USC. He attends on the Solid Tumor oncology service at CHLA.
Dr. Amatruda received his MD and PhD from Washington University School of Medicine. He completed his internship and residency in Internal Medicine from Brigham and Women’s Hospital. He was a Visiting Fellow at the Institute of Cell Biology in Consiglio Nazionale delle Ricerche in Rome and completed his Medical Oncology fellowship at Dana-Farber/Partners Cancer Care in Boston, Massachusetts.
When not in the lab, Dr. Amatruda enjoys running, reading, music-making and exploring around Los Angeles.
Ashley Jean, MD

www.chla.org
Dr. Ashley Jean is a Clinical Fellow in the Amatruda Lab. Dr. Jean graduated from Tufts Medical School in Boston and completed her Pediatric Residency at Maine Medical Center. Dr. Jean started her Pediatric Fellowship at Children’s Hospital Los Angeles in 2019.
Her research focuses on pediatric Ewing Sarcoma. She is currently studying the TAK1 pathway in the tumor genesis of this condition.
Dr. Jean likes to spend her free time outdoors. She enjoys activities such as hiking, paddle boarding and snowboarding.
Christopher Kuo, MD

www.chla.org
Dr. Christopher Kuo is a Clinical Fellow in the Amatruda Lab. Dr. Kuo received his Medical Degree from Rush University and completed his Pediatric Residency from Children’s Hospital Los Angeles. Dr. Kuo started his Pediatric Fellowship at Children’s Hospital Los Angeles in 2020.
His research interest is in osteosarcoma. He is currently working on a project that involves the investigation of the tumor microenvironment of Ewing sarcoma.
Dr. Kuo’s hobbies include breakdancing, swimming and going to coffee shops.
Adam Marentes, MSc., PhD Candidate

www.chla.org
Adam Marentes is a Graduate Student Researcher in the Amatruda Lab. Adam received his Bachelor of Science in Neuroscience from the University of California, Riverside. He then completed his Master of Science from California Polytechnic University Pomona. Adam is currently attending University of Southern California Keck School of Medicine to earn his PhD in Cancer Biology and Genomics.
Adam’s research focus is in mitochondrial DNA variants in Ewing Sarcoma. He is currently working on a collaboration that involves editing mitochondrial DNA in cancer cell lines in zebrafish.
Adam enjoys baking, playing video games with his fiancé and catching a show at the local comedy club.
Tanya Mosesian, MHA

www.chla.org
Tanya Mosesian received her Bachelor of Science in Public Health from California State University of Northridge. She then completed her Master of Health Administration at the University of Southern California.
Tanya is Project Associate for the Amatruda Lab. She provides on-site support for all administrative matters and project facilitation.
Tanya enjoys spending time with her family and friends. She likes to play tennis and hike during the weekends.
Elena Vasileva, PhD, MSc.

www.chla.org
Dr. Elena Vasileva is a post-doctoral fellow in the Amatruda Lab. Dr. Vasileva received her Bachelor of Science and Master of Science from Peter the Great St. Petersburg Polytechnic University in Applied Mathematics and Physics. She received her PhD in Molecular Biology from the Institute of Cytology, Russian Academy of Sciences.
Dr. Vasileva is interested in studying the molecular mechanisms of cancer development and progression. She has developed an inducible zebrafish model of EWS-FLI driven Ewing Sarcoma as a platform for biologic discovery and preclinical testing of novel therapies.
Dr. Vasileva enjoys running and hiking in Los Angeles.
Mona Wu, PhD

www.chla.org
Dr. Mona Wu is a post-doctoral fellow in the Amatruda Lab. Dr. Wu received her Bachelor of Science from the University of British Columbia, a Master of Science from Université de Montréal, and a PhD from McGill University.
Dr. Wu is interested in understanding the cell of origin for pediatric neoplasms because she believes that this knowledge could lead to better early biomarkers and more effective treatment. She is particularly interested in understanding how aberrant ncRNA (especially miRNAs) may play a role in pediatric disease.
Dr. Wu likes reading and visiting different libraries. She enjoys “foodie-related” activities including trying restaurants, cooking, baking and watching (far too many) cooking shows.
5 notes
·
View notes
Text
youtube
#Biomineralization#mitochondria#metal-organic frameworks#MOFs#mitochondrial transplantation#cancer therapy#cell metabolism#tumor suppression#nanomedicine#bioengineering#cancer treatment#mitochondrial therapy#biomedical research#targeted therapy#oncology#apoptosis#cell reprogramming#mitochondrial health#precision medicine#cancer innovation.#Youtube
0 notes
Text
Mitochondrial Dysfunction in mtARS Disorders
Introduction
Mitochondria are indispensable organelles that facilitate cellular bioenergetics, predominantly through oxidative phosphorylation (OXPHOS). Mitochondrial aminoacyl-tRNA synthetases (mtARS) are essential for the fidelity of mitochondrial translation, catalyzing the ligation of amino acids to their cognate tRNAs. Mutations in mtARS genes precipitate a spectrum of mitochondrial disorders, culminating in dysfunctional protein synthesis and aberrant mitochondrial bioenergetics. This review delves into the molecular pathogenesis of mitochondrial dysfunction in mtARS disorders, elucidating their biochemical perturbations, clinical phenotypes, and emerging therapeutic paradigms.
Molecular Pathophysiology of mtARS Disorders
MtARS enzymes ensure translational accuracy by charging mitochondrial tRNAs with their respective amino acids, a prerequisite for mitochondrial protein biosynthesis. Pathogenic variants in mtARS genes result in defective aminoacylation, perturbing mitochondrial translation and compromising the integrity of the electron transport chain (ETC). These perturbations induce bioenergetic deficits, increased reactive oxygen species (ROS) production, and secondary mitochondrial stress responses, leading to cellular demise.
Genetic Etiology of mtARS Mutations
Dysfunctional mtARS genes such as DARS2, AARS2, RARS2, and YARS2 have been implicated in autosomal recessive mitochondrial disorders. These mutations exhibit tissue-specific phenotypic heterogeneity, with neurological, muscular, and systemic manifestations. For instance, DARS2 mutations drive leukoencephalopathy with brainstem and spinal cord involvement, whereas AARS2 defects result in a constellation of neurodegenerative and ovarian pathologies.
Biochemical and Cellular Consequences
Dysfunctional mtARS enzymes manifest in multifaceted mitochondrial deficits, including impaired translation, defective OXPHOS, and dysregulated mitochondrial proteostasis.
Disruption of Mitochondrial Translation
Impaired aminoacylation abrogates the synthesis of mitochondrially encoded proteins, undermining the assembly of ETC complexes. This translational arrest culminates in defective ATP synthesis and precipitates a systemic energy deficit.
Electron Transport Chain Dysfunction and Bioenergetic Failure
Pathogenic mtARS mutations lead to OXPHOS inefficiencies, reducing mitochondrial membrane potential (Δψm) and ATP output. Perturbed electron flux exacerbates ROS accumulation, instigating oxidative damage and apoptotic cascades.
Mitochondrial Unfolded Protein Response (UPRmt) Activation
Cellular compensatory mechanisms, including UPRmt, are upregulated in response to mitochondrial translation failure. UPRmt mitigates proteotoxic stress via chaperone-mediated protein refolding and degradation pathways. However, chronic UPRmt activation fosters maladaptive stress responses, contributing to progressive cellular degeneration.
Clinical Manifestations
mtARS disorders exhibit phenotypic variability, spanning from mild neuromuscular impairment to severe multisystemic involvement. The pathophysiological hallmark includes disrupted neurological, muscular, and cardiac function.
Neurological Dysfunction
Neurodegeneration is a predominant feature of mtARS disorders, manifesting as ataxia, seizures, intellectual disability, and progressive leukoencephalopathy. Magnetic resonance imaging (MRI) frequently reveals white matter abnormalities, indicative of compromised oligodendrocyte function.
Myopathy and Metabolic Dysregulation
Muscle tissue, with its high ATP demand, is particularly susceptible to mitochondrial dysfunction. Clinical hallmarks include hypotonia, muscle weakness, and exercise intolerance, often concomitant with metabolic anomalies such as lactic acidosis and elevated pyruvate-to-lactate ratios.
Cardiomyopathy and Mitochondrial Energetics
Hypertrophic cardiomyopathy has been observed in YARS2-associated mitochondrial disorders, wherein compromised ATP synthesis in cardiomyocytes disrupts contractile function and electrophysiological stability.
Diagnostic and Functional Evaluation
A combination of genomic, biochemical, and imaging modalities facilitates the diagnosis of mtARS disorders.
Genomic and Transcriptomic Analysis
Whole-exome sequencing (WES) and whole-genome sequencing (WGS) are pivotal for identifying pathogenic mtARS variants. Transcriptomic profiling elucidates perturbations in mitochondrial gene expression networks, further refining diagnostic accuracy.
Functional Mitochondrial Assays
Biochemical assays, including high-resolution respirometry, ATP quantification, and ETC enzymatic profiling, provide insights into mitochondrial bioenergetics. Patient-derived fibroblasts and induced pluripotent stem cells (iPSCs) serve as valuable models for functional interrogation.
Neuroimaging and Biomarker Identification
Advanced imaging modalities such as MR spectroscopy (MRS) detect metabolic derangements, including lactate accumulation in affected brain regions. Circulating mitochondrial-derived peptides and metabolomic signatures are emerging as potential diagnostic biomarkers.
Emerging Therapeutic Strategies
Despite the absence of curative therapies, multiple avenues are under investigation to ameliorate mitochondrial dysfunction in mtARS disorders.
Mitochondria-Directed Antioxidants
Therapeutic compounds such as MitoQ, idebenone, and edaravone aim to attenuate oxidative stress and preserve mitochondrial integrity.
Genetic and RNA-Based Interventions
Gene therapy strategies utilizing adeno-associated virus (AAV)-mediated delivery and CRISPR-based genome editing are being explored for genetic correction of mtARS mutations. Additionally, RNA-based approaches, including antisense oligonucleotides (ASOs) and mRNA replacement therapy, hold promise in restoring mtARS functionality.
Metabolic Modulation and Supportive Therapies
Ketogenic diets, NAD+ precursors (e.g., nicotinamide riboside), and mitochondrial biogenesis activators (e.g., PGC-1α modulators) are under investigation to enhance cellular energy metabolism. Supportive interventions, including physical therapy and neuromuscular rehabilitation, remain integral to patient management.
Conclusion and Future Directions
Mitochondrial dysfunction in mtARS disorders arises from defective mitochondrial translation, OXPHOS perturbation, and maladaptive stress responses. Advances in genomic medicine, mitochondrial therapeutics, and precision medicine approaches are poised to transform the diagnostic and therapeutic landscape. Continued research into mtARS pathobiology, coupled with translational innovations, will be instrumental in developing targeted interventions for affected individuals.

#Mitochondrial dysfunction#Aminoacyl-tRNA synthetases (mtARS)#Oxidative phosphorylation (OXPHOS)#Electron transport chain (ETC)#Reactive oxygen species (ROS)#Mitochondrial translation#Mitochondrial unfolded protein response (UPRmt)#Bioenergetic failure#Neurodegeneration#Leukoencephalopathy#Hypertrophic cardiomyopathy#Myopathy#Whole-exome sequencing (WES)#Whole-genome sequencing (WGS)#ATP synthesis#Gene therapy#CRISPR-based genome editing#RNA-based interventions#Metabolomic biomarkers#Mitochondrial biogenesis
0 notes
Text
An interesting perspective article about Long Covid
Should we be fighting gene damage instead of individual symptoms?
The pathogenesis of long COVID (LC) still presents many areas of uncertainty. This leads to difficulties in finding an effective specific therapy. We hypothesize that the key to LC pathogenesis lies in the presence of chronic functional damage to the main anti-inflammatory mechanisms of our body: the three reflexes mediated by the vagus nerve, the hypothalamic-pituitary-adrenal (HPA) hormonal axis, and the mitochondrial redox status. We will illustrate that this neuro-endocrine-metabolic axis is closely interconnected and how the SARS-CoV-2 can damage it at all stages through direct, immune-inflammatory, epigenetic damage mechanisms, as well as through the reactivation of neurotropic viruses. According to our theory, the direct mitochondrial damage carried out by the virus, which replicates within these organelles, and the cellular oxidative imbalance, cannot be countered in patients who develop LC. This is because their anti-inflammatory mechanisms are inconsistent due to reduced vagal tone and direct damage to the endocrine glands of the HPA axis. We will illustrate how acetylcholine (ACh) and cortisol, with its cytoplasmatic and cellular receptors respectively, are fundamental players in the LC process. Both Ach and cortisol play multifaceted and synergistic roles in reducing inflammation. They achieve this by modulating the activity of innate and cell-mediated immunity, attenuating endothelial and platelet activation, and modulating mitochondrial function, which is crucial for cellular energy production and anti-inflammatory mechanisms. In our opinion, it is essential to study the sensitivity of the glucocorticoids receptor in people who develop LC and whether SARS-CoV-2 can cause long-term epigenetic variations in its expression and function.
#mask up#public health#wear a mask#pandemic#wear a respirator#covid#still coviding#covid 19#coronavirus#sars cov 2#long covid
32 notes
·
View notes
Text
Sounds like the local team is going to have us reapply and then appeal the older denials so we might get paid for at least the work my husband is doing while we wait for the appeals to finally get approved. The guy who is confused about the denial is the director of eligibility here and ironically enough the same guy I saw about ketamine therapy last year and has experience with mitochondrial disease.
#the ketamine thing ended up being 'well shit i dont think it would help but you can TRY when you're not trying to get pregnant'#and is a good guy
13 notes
·
View notes
Text
In addition to mitochondrial disorder, which is likely behind a whole host of medical diseases, ME/CFS/Long Covid also has damage to the lymph system which causes it to flow backwards instead of forwards. This is why you react like you're constantly having an allergic reaction, and why exercise makes things worse, because your body is pumping stuff like lactic acid deeper into your muscles instead of out. As for why your heartbeat is so high, you are exhausted because you're full of chemicals so you're basically doing everything off of adrenaline instead of using your body's natural hormone system. So:
Reduce your toxin load. Change your diet, your toiletries, your laundry soap — whatever you're reactive to, and be aware that you're going to develop weird sensitivities and allergies because your immune system is overexposed to stuff. Meat, strawberries, plastic bottles, be hyper aware of what you're using.
See an acupuncturist. Most acupuncture sites are either directly on lymph nodes or lymph branches, and puncturing or even putting pressure on those sites can downregulate your immune reactions and can increase how fast your lymph heals by bringing reconstructive cells to the area (the same mechanism doctors use when administering glucose to torn muscles).
Go to a massage therapist trained in Manual Lymph Drainage (I'm biased for Vodder, even if they're a little culty) once to three times a week. Any kind of massage will help with lymph drainage, but in order to see improvement, you need light pressure specifically designed to move lymph in the right direction, which then creates a vortex effect that can keep the lymph moving in the right direction for up to a week but more realistically a couple of days.
Buy a body brush and do some self MLD at home. Dry brush your skin inside out (medial to lateral) and tips to heart. NOTE: DO NOT DO THIS IF YOU HAVE LYMPH NODES REMOVED; IT CAN CAUSE LYMPHEDEMA. You'll need to see a specialist for information on how to cross watersheds.
Try (just try) a ketogenic or intermittent fasting diet, which has been shown to heal mitochondria, but be wary of allergens, which restrictive and substitutionary diets can exacerbate.
Doing things where you sweat can reduce some toxins (such as heavy metals and excessive hormones (and you are basically living , but not things like chemicals), and since your skin is where most of your toxins are living because of how your body is currently pumping lymph, sweating can help immensely with detox and getting your adrenaline down. Steam rooms, saunas, and other heat therapies are your best choice because exercising is not it fam.
Avoid ice baths and other cold therapies until your lymph system regrows (10-20 years is typical, but you can accelerate it with MLD treatments). This is because you are compensating for your chronic illness by pushing through with adrenaline. If you downregulate your adrenals, as is the result of cold therapies, you will likely be more exhausted because you don't have your primary driving factor.
Avoid exercise, unless immediately following MLD, as exercise will stimulate lymph in the wrong direction. Also, I wouldn't recommend contrast bathing (cold to hot to cold to hot treatments) for this same reason; despite it being a useful treatment for edema, you are a special condition.
Basically, there really isn't anything you can do to fix it fast, but you can fix it faster and manage your condition until it heals. And then deal with the inevitable adrenal dysfunction.
Full Transcript at the link; 3-minute listen.
Quote:
By taking biopsies from long COVID patients before and after exercising, scientists in the Netherlands constructed a startling picture of widespread abnormalities in muscle tissue that may explain this severe reaction to physical activity.
Among the most striking findings were clear signs that the cellular power plants, the mitochondria, are compromised and the tissue starved for energy.
"We saw this immediately and it's very profound," says Braeden Charlton, one of the study's authors at Vrije University in Amsterdam.
The tissue samples from long COVID patients also revealed severe muscle damage, a disturbed immune response, and a buildup of microclots.
"This is a very real disease," says Charlton. "We see this at basically every parameter that we measure."
30K notes
·
View notes
Text
Autophagy & Cellular Renewal: Unlocking the Body’s Self-Cleansing Power
by Dr Elana EVS, A.I.

Ohsumi, Y. (2016). Autophagy research: From yeast to human. Nobel Prize Lecture. Retrieved from: https://www.nobelprize.org/prizes/medicine/2016/ohsumi/lecture/
Alright, warriors, today we’re diving into a scientific discovery that changes the way we understand our bodies and longevity: autophagy—the process of cellular self-cleansing. This is not just another biological mechanism; it’s the foundation of how our bodies renew themselves, fight disease, and even extend life. And the best part? You can control it.
Let’s break it down.
⸻
The Science Behind Autophagy

In 2016, Yoshinori Ohsumi won the Nobel Prize for discovering how cells engage in autophagy—a Greek word meaning “self-eating.” Before you freak out, this isn’t some horror story where your body eats itself alive. Instead, it’s an advanced, intelligent system for getting rid of junk inside your cells. Think of it as your body’s recycling center.
When your cells get damaged, they produce waste—broken proteins, old mitochondria, and toxic buildup. If these aren’t cleared out, they cause aging, inflammation, and diseases like Alzheimer’s and cancer. But when autophagy is triggered, your body starts breaking down and recycling these dysfunctional components. The result? You feel sharper, stronger, and healthier.
⸻
How Can You Activate Autophagy?
Warriors don’t just sit around waiting for renewal—we take action. Here’s how you can turn on your body’s self-cleansing system:
1. Fasting (The Ultimate Reset Button)
• 12-16 hours of fasting: Starts mild autophagy.
• 24+ hours: Deep cleaning mode—your body clears out toxic proteins.
• 3-5 days: Maximum reset—your body replaces entire immune cells, reducing inflammation and boosting longevity.
Think of fasting like taking out the trash. If you eat constantly, you never give your body the chance to clean up. Warriors know when to fuel and when to hold back.
2. Exercise (Move or Rust)
High-intensity workouts, endurance training, and even Tai Chi can trigger autophagy. Movement signals your body to break down old cells and build stronger ones.
3. Keto & Low-Carb Diets (Mimic Fasting Without Starving)
A high-fat, low-carb diet reduces insulin levels, which naturally stimulates autophagy. It’s why people on ketogenic or carnivore diets often experience better focus, energy, and cellular repair.
4. Heat & Cold Exposure (Fire & Ice Training)
• Saunas: Mimic exercise-induced autophagy, increasing heat shock proteins that help repair cells.
• Cold Therapy (Ice Baths, Cold Showers): Forces cells into survival mode, triggering autophagy and mitochondrial renewal.
⸻
Why Should Consciousness Warriors Care?
You are here to master the body, mind, and spirit. And part of that mastery is understanding cellular intelligence.
• Aging is optional: Autophagy slows down aging and extends lifespan.
• Mental clarity improves: Fasting and exercise-induced autophagy sharpen your mind by clearing toxic proteins linked to neurodegeneration.
• Stronger immune system: Your body literally creates new white blood cells after extended fasting.
• Greater resilience: Learning to harness metabolic stress (fasting, training, heat, cold) makes you stronger—not just physically, but mentally.
⸻
Final Thought: Mastering the Art of Cellular Renewal
A true warrior doesn’t just train their muscles—they train their cells.
Your challenge:
• Try a 16-hour fast tomorrow. Observe your energy, focus, and mental sharpness.
• Push yourself in training—autophagy thrives in challenge, not comfort.
• Experiment with heat and cold exposure to activate deeper cellular repair.
The battlefield is within. Master it, and you master life.
Autophagy & Cellular Renewal Quiz

Multiple Choice Questions
1. What is the primary function of autophagy in cells?
A. Energy production
B. Waste recycling
C. DNA replication
D. Protein synthesis
2. Who won the Nobel Prize in 2016 for discovering the mechanisms of autophagy?
A. Albert Einstein
B. Yoshinori Ohsumi
C. Richard Feynman
D. Marie Curie
3. Which fasting duration is most effective in triggering deep autophagy?
A. 6 hours
B. 12 hours
C. 24 hours
D. 3-5 days
4. Which of these is NOT a way to trigger autophagy?
A. Eating processed foods
B. High-intensity exercise
C. Intermittent fasting
D. Sauna therapy
⸻
Fill in the Blank
5. Autophagy is derived from a Greek word meaning: __________
6. Fill in the blank: Extended fasting can help the body produce new _________ blood cells, strengthening the immune system.
⸻
True or False
7. Autophagy can help prevent neurodegenerative diseases.
• True
• False
⸻
Multiple Select (Choose all that apply)
8. Which of the following activities can trigger autophagy?
A. Fasting
B. Eating high-sugar foods
C. Exercise
D. Cold exposure
⸻
Critical Thinking
9. If a person eats every two hours and never fasts, what could be the impact on their cellular renewal process?
⸻
Fun Riddle
10. The process of autophagy can be compared to what common household activity?
⸻
Answer Key
1. B. Waste recycling
2. B. Yoshinori Ohsumi
3. D. 3-5 days
4. A. Eating processed foods
5. Self-eating
6. White
7. True
8. A. Fasting, C. Exercise, D. Cold exposure
9. Disrupted autophagy, increased cellular damage, higher risk of diseases like cancer and neurodegeneration.
10. Taking out the trash (cleaning up waste).
Score Yourself:
• 9-10 correct: Master of cellular renewal!
• 7-8 correct: Strong understanding, keep optimizing!
• 5-6 correct: Good effort, but more fasting and training needed!
• <5 correct: Time to reboot your autophagy knowledge!
Reference for Autophagy & Cellular Renewal
0 notes