
Don't wanna be here? Send us removal request.
Statistics
We looked inside some of the posts by blueoaknx and here's what we found interesting.
Average Info
Notes Per Post
1
Likes Per Post
1
Reblog Per Post
0
Reply Per Post
0
Time Between Posts
12 days
Number of Posts By Type
Text
17
Last Seen Tumblr Blogs





Fun Fact
The Tumblr app for Google Glass was released on May 16, 2013.
Text
The Role of Mitochondria in Prader–Willi Syndrome
1. Introduction
Prader–Willi Syndrome (PWS) is a rare genetic disorder resulting from the lack of expression of paternally inherited genes in the 15q11–q13 chromosomal region. Clinically, it is characterized by neonatal hypotonia, hyperphagia, obesity, short stature, cognitive impairment, hypogonadism, and behavioral issues. Historically, these features have been attributed to hypothalamic dysfunction. However, recent research highlights a significant role of mitochondrial dysfunction in the metabolic and neuromuscular symptoms of PWS.
2. Mitochondrial Function and Its Systemic Relevance
Mitochondria are cellular organelles essential for energy production through oxidative phosphorylation (OXPHOS). They also regulate reactive oxygen species (ROS) generation, calcium signaling, and apoptosis. In energy-demanding tissues such as brain and muscle, mitochondrial integrity is vital. Any impairment in mitochondrial function disrupts cellular energy metabolism, often resulting in clinical features seen in syndromes like PWS.
3. Bioenergetic Deficits in PWS
Patients with PWS exhibit symptoms like muscle weakness, reduced endurance, and fatigue—all suggestive of compromised mitochondrial energy production. Cellular studies on fibroblasts derived from PWS individuals have shown decreased basal respiration, reduced ATP production, and limited spare respiratory capacity. These deficits indicate impaired mitochondrial oxidative phosphorylation and diminished cellular energy reserves.
4. Electron Transport Chain Abnormalities
Specific defects in the electron transport chain (ETC), particularly in Complex I, have been reported in PWS. Complex I initiates the ETC by transferring electrons from NADH to ubiquinone. Defects in Complex I result in lower ATP generation and an increase in ROS. The resultant oxidative stress can damage mitochondrial DNA, lipids, and proteins, further impairing mitochondrial function and exacerbating clinical symptoms.
5. Coenzyme Q10 Deficiency
Coenzyme Q10 (CoQ10) is a lipid-soluble molecule vital for electron transport between Complexes I/II and III. It also acts as an antioxidant, protecting membranes and cellular structures from oxidative damage. In individuals with PWS, CoQ10 levels are often significantly lower than in the general population. This deficiency disrupts electron flow, reduces ATP synthesis, and increases oxidative stress. Clinically, CoQ10 deficiency may contribute to hypotonia, poor endurance, and delayed developmental milestones in PWS patients.
6. Fatty Acid Oxidation and Acylcarnitine Abnormalities
In PWS, metabolic profiling has revealed elevated acylcarnitine levels, particularly medium- and short-chain species. These findings suggest a disruption in fatty acid β-oxidation, a key mitochondrial process. Accumulated acylcarnitines are indicative of incomplete fatty acid utilization, which may stem from defective carnitine transport or mitochondrial enzyme activity. As fatty acids are critical energy substrates during fasting and exercise, their impaired oxidation contributes to energy failure and obesity in PWS.
7. Carnitine Deficiency and Transport Impairment
Carnitine is essential for the transport of long-chain fatty acids into mitochondria for β-oxidation. Some studies have reported reduced serum carnitine levels in individuals with PWS, especially in infants and young children. Carnitine deficiency may result from reduced intake, increased renal losses, or altered synthesis. Supplementation with carnitine has been associated with improvements in muscle tone and energy levels in some cases, suggesting its therapeutic potential.
8. Gene Expression and Mitochondrial Regulation
PWS results from the loss of paternal expression of genes in the 15q11–q13 region, including small nucleolar RNAs (snoRNAs) and non-coding RNAs involved in RNA processing and regulation. Transcriptomic studies in mouse models have shown dysregulation of genes associated with mitochondrial function, including those involved in ribosomal assembly, fatty acid metabolism, and oxidative phosphorylation. These molecular alterations reinforce the hypothesis that mitochondrial dysfunction is a primary contributor to the PWS phenotype.
9. Structural Mitochondrial Alterations
Electron microscopy studies in animal models of PWS have demonstrated mitochondrial structural abnormalities, including swelling, disorganized cristae, and altered mitochondrial number. These findings correlate with decreased efficiency of oxidative metabolism and increased oxidative damage. Mitochondrial remodeling in cardiac, neural, and skeletal muscle tissues may underlie systemic features such as cardiomyopathy, cognitive deficits, and fatigue.
10. Therapeutic Implications
Understanding mitochondrial dysfunction in PWS opens the door to targeted therapies. The following strategies are under consideration:
Coenzyme Q10 Supplementation: Administered to enhance electron transport and reduce oxidative stress. Anecdotal reports have shown improved motor function and alertness in children receiving CoQ10.
Carnitine Therapy: May support fatty acid transport and improve energy production. Used in cases with documented deficiency or fatigue.
Antioxidants: Agents such as alpha-lipoic acid, vitamin E, or NAC might mitigate ROS-related damage and preserve mitochondrial integrity.
Mitochondrial Biogenesis Enhancers: Agents that stimulate mitochondrial replication and function, such as PGC-1α activators, are under investigation.
Metabolic Monitoring: Regular assessment of acylcarnitine profiles, lactate, and oxidative stress markers can help personalize treatment.
11. Future Directions
To advance clinical care for PWS, several research priorities have emerged:
Controlled Clinical Trials: Rigorous evaluation of CoQ10 and carnitine supplementation is needed to assess efficacy and safety.
Multi-Tissue Profiling: Comprehensive mitochondrial function studies in muscle, brain, liver, and adipose tissues will clarify tissue-specific vulnerabilities.
Genotype–Phenotype Correlation: Understanding how specific genetic deletions affect mitochondrial pathways can guide personalized interventions.
Biomarker Development: Identifying mitochondrial biomarkers in blood or urine could enable early detection of dysfunction and monitoring of treatment response.
12. Conclusion
While traditionally attributed to hypothalamic dysfunction, Prader–Willi syndrome also involves systemic mitochondrial impairment. Defects in energy metabolism, fatty acid oxidation, and antioxidant defense converge to produce many of the syndrome's characteristic features. Recognition of mitochondrial involvement in PWS pathophysiology has the potential to refine diagnosis, improve symptom management, and inspire new therapeutic avenues. Future research integrating genomics, bioenergetics, and clinical studies will be essential in translating this understanding into effective patient care.
#Prader–Willi Syndrome#Mitochondrial dysfunction#Oxidative phosphorylation (OXPHOS)#Coenzyme Q10 (CoQ10)#Carnitine deficiency#Electron transport chain#Complex I activity#Fatty acid oxidation#Acylcarnitines#Reactive oxygen species (ROS)#Mitochondrial bioenergetics#Energy metabolism#Hypotonia in PWS#Metabolic profiling#Mitochondrial gene expression#Mitochondrial structure#Mitochondrial therapy#Antioxidant supplementation#Mitochondrial biogenesis#Neuromuscular symptoms PWS
0 notes
Text
The Role of Mitochondria in Amyotrophic Lateral Sclerosis (ALS)

Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterized by the progressive degeneration of upper and lower motor neurons. Despite extensive research, the etiology of ALS remains multifactorial and incompletely understood. Among the cellular organelles implicated in ALS pathogenesis, mitochondria stand out as central mediators of neurodegeneration due to their pivotal roles in ATP production, calcium homeostasis, and apoptosis regulation. Mitochondrial dysfunction is not merely a secondary feature of dying neurons in ALS; accumulating evidence suggests it plays a primary and causative role in disease progression.
Mitochondrial Bioenergetics in ALS
Mitochondria are indispensable for neuronal survival due to their role in oxidative phosphorylation (OXPHOS), the process that generates over 90% of cellular ATP. In ALS, impairments in mitochondrial respiration are evident across multiple models, including post-mortem human spinal cord tissue, motor neurons derived from induced pluripotent stem cells (iPSCs), and transgenic mouse models harboring ALS-associated mutations (e.g., SOD1, TDP-43, FUS, C9orf72).
Notably, enzymatic activity of complexes I and IV of the electron transport chain is reduced in ALS, correlating with early energy deficits in motor neurons. These neurons are especially vulnerable due to their long axons and high metabolic demand. Impaired ATP synthesis not only compromises synaptic transmission but also disrupts axonal transport and the maintenance of ion gradients, rendering neurons more susceptible to excitotoxicity and death.
Oxidative Stress and Reactive Oxygen Species (ROS)
Mitochondria are the principal source of reactive oxygen species as byproducts of respiration. While low levels of ROS serve signaling functions, excessive ROS can damage lipids, proteins, and DNA. ALS is associated with increased oxidative stress, evidenced by elevated markers of lipid peroxidation and protein carbonylation in cerebrospinal fluid and nervous tissue.
Mutations in SOD1, one of the first discovered ALS-associated genes, further underscore the connection between oxidative stress and mitochondrial dysfunction. SOD1 normally detoxifies superoxide radicals. Mutant SOD1 misfolds and aggregates within mitochondria, particularly in the intermembrane space, impairing mitochondrial integrity and exacerbating ROS production. This establishes a vicious cycle wherein dysfunctional mitochondria produce more ROS, leading to further mitochondrial damage.
Mitochondrial Dynamics: Fission, Fusion, and Transport
Mitochondria are dynamic organelles that constantly undergo fission and fusion, processes necessary for maintaining mitochondrial function and distribution. These dynamics are disrupted in ALS. Studies have demonstrated increased mitochondrial fragmentation in motor neurons from ALS models, often linked to elevated activity of fission proteins such as DRP1 and downregulation of fusion mediators like MFN2 and OPA1.
Additionally, mitochondrial transport along axons is impaired in ALS. Mitochondria must be trafficked to sites of high energy demand, including synaptic terminals. Mutant SOD1, TDP-43, and FUS have all been implicated in disrupting the interaction between mitochondria and motor proteins such as kinesin and dynein. This disruption leads to a depletion of functional mitochondria at distal axonal sites, contributing to synaptic failure and distal axonopathy, hallmarks of ALS pathology.
Calcium Dysregulation and Excitotoxicity
Motor neurons in ALS are particularly sensitive to calcium dysregulation, and mitochondria play a vital role in buffering intracellular calcium. Under pathological conditions, mitochondria in ALS exhibit reduced calcium uptake capacity. This dysfunction is partly due to depolarized mitochondrial membrane potential and possibly due to defective interactions at mitochondria-associated membranes (MAMs), where calcium is transferred from the endoplasmic reticulum (ER) to mitochondria.
The result is excessive cytosolic calcium, which, when combined with increased glutamate signaling, leads to excitotoxicity. This not only activates calcium-dependent proteases and phospholipases but also triggers the opening of the mitochondrial permeability transition pore (mPTP), a catastrophic event that leads to mitochondrial swelling, rupture, and the release of pro-apoptotic factors.
Apoptosis and Mitochondrial Permeability
The intrinsic pathway of apoptosis is closely regulated by mitochondrial integrity. In ALS, numerous studies have identified mitochondrial-mediated apoptosis as a significant contributor to motor neuron death. Proteins such as Bax and Bak insert into the mitochondrial outer membrane, promoting cytochrome c release and subsequent caspase-3 activation. Elevated levels of cleaved caspase-9 and caspase-3 have been reported in ALS patient tissue and transgenic models.
Moreover, persistent mitochondrial stress leads to chronic opening of the mPTP, a key event that commits the cell to death. This process is further exacerbated by oxidative stress, calcium overload, and the presence of misfolded proteins, all of which are abundant in ALS-affected neurons.
Defective Mitophagy and Quality Control
Quality control of mitochondria is essential to neuronal homeostasis. Damaged mitochondria are normally removed by mitophagy, a specialized form of autophagy. In ALS, mitophagy is often impaired. Mutations in genes like OPTN, TBK1, and VCP—all associated with familial ALS—directly interfere with mitophagy pathways. These proteins are responsible for tagging damaged mitochondria for autophagic clearance via ubiquitination and recruitment of autophagic machinery.
When mitophagy is compromised, dysfunctional mitochondria accumulate, leading to sustained oxidative damage, ATP deficiency, and activation of cell death pathways. This accumulation also leads to inflammation, as damaged mitochondria can release mitochondrial DNA (mtDNA) and other damage-associated molecular patterns (DAMPs) that activate innate immune responses.
Therapeutic Implications
Given the central role of mitochondria in ALS, numerous therapeutic strategies aim to restore mitochondrial function or prevent their dysfunction. Antioxidants such as coenzyme Q10, edaravone, and idebenone have been tested, with edaravone gaining limited clinical approval for slowing functional decline.
Agents targeting mitochondrial dynamics (e.g., DRP1 inhibitors), enhancing mitophagy (e.g., urolithin A), or stabilizing mitochondrial membranes are under preclinical and clinical investigation. Additionally, metabolic modulators that shift energy production away from oxidative phosphorylation (e.g., ketogenic diets or dichloroacetate) show promise in experimental models.
However, the translation of mitochondrial-targeted therapies into effective clinical treatments remains challenging. This is due in part to the heterogeneity of ALS, the complexity of mitochondrial biology, and the difficulty in delivering drugs across the blood-brain barrier in therapeutically relevant concentrations.
Conclusion
Mitochondria are at the intersection of multiple pathogenic pathways in ALS, including energy failure, oxidative stress, calcium overload, impaired dynamics, and defective quality control. Far from being mere bystanders, these organelles are deeply implicated in both initiating and propagating motor neuron degeneration. Future advances in ALS therapy will likely depend on a deeper understanding of mitochondrial biology and the development of strategies that can restore or preserve mitochondrial health in vulnerable neuronal populations.
#Amyotrophic Lateral Sclerosis (ALS)#Mitochondrial dysfunction#Motor neuron degeneration#Oxidative stress#Reactive oxygen species (ROS)#SOD1 mutation#Mitochondrial bioenergetics#Electron transport chain#ATP production#Mitochondrial dynamics#Mitochondrial fission and fusion#Axonal transport#Calcium dysregulation#Excitotoxicity#Mitochondrial apoptosis#Mitophagy#Mitochondrial permeability transition pore (mPTP)#Mitochondria-associated membranes (MAMs)#Neurodegeneration
0 notes
Text
The Role of Mitochondria in Autism Spectrum Disorder

Introduction
Autism Spectrum Disorder (ASD) is a neurodevelopmental condition defined by difficulties in communication, social interaction, and the presence of repetitive behaviors. While ASD's exact origins remain complex and multifaceted, growing research highlights mitochondrial dysfunction as a key biological contributor. Mitochondria are vital organelles responsible for generating cellular energy and maintaining homeostasis, particularly in energy-demanding organs like the brain. Impairments in mitochondrial function can significantly disrupt neural development and have been increasingly observed in individuals with ASD. This article delves into the role of mitochondria in ASD, exploring evidence of mitochondrial abnormalities and their implications for understanding and treating this condition.
Mitochondrial Function and Brain Development
Mitochondria produce adenosine triphosphate (ATP), the energy currency of cells, through a process known as oxidative phosphorylation. This energy production is essential for many brain processes including neurotransmission, synaptic plasticity, and cellular repair. Besides energy generation, mitochondria are also involved in regulating calcium levels, producing reactive oxygen species (ROS), and controlling apoptosis (programmed cell death). All these functions are particularly important during early brain development when neurons are rapidly forming connections and networks.
Mitochondrial Dysfunction in Autism
Abnormal Biochemical Profiles Numerous studies have detected elevated levels of lactate, pyruvate, and alanine in the blood and cerebrospinal fluid of individuals with ASD. These findings suggest a disruption in mitochondrial energy metabolism. Abnormal lactate-to-pyruvate ratios, for example, point to oxidative phosphorylation inefficiencies, implying that mitochondria in ASD-affected individuals are not functioning optimally.
Increased Oxidative Stress Mitochondria are both producers and targets of ROS, and when not properly regulated, ROS can damage DNA, proteins, and lipids. In individuals with ASD, elevated markers of oxidative stress and reduced levels of antioxidants such as glutathione have been reported. This imbalance can contribute to neural inflammation and impair neurodevelopment, possibly exacerbating core ASD symptoms.
Genetic Abnormalities in Mitochondrial DNA Some individuals with ASD exhibit mutations or deletions in mitochondrial DNA (mtDNA). Since mtDNA is crucial for the normal function of the electron transport chain—essential for ATP production—such mutations can compromise cellular energy availability. Furthermore, mitochondrial diseases, which often involve mtDNA mutations, frequently present with neurodevelopmental symptoms overlapping with those of ASD.
Mitochondrial Dynamics and Quality Control Mitochondrial health depends on processes such as fission, fusion, and mitophagy (the removal of damaged mitochondria). In ASD, studies have observed altered expressions of genes involved in these dynamic processes. Imbalances in mitochondrial fission and fusion can lead to dysfunctional mitochondria accumulating in neurons, impairing their function and survival.
Impact on Synaptic Function Efficient synaptic transmission relies heavily on mitochondrial energy. Mitochondria located at synapses help regulate calcium signaling and provide the necessary ATP for neurotransmitter release. Mitochondrial dysfunction may therefore contribute to the synaptic abnormalities frequently observed in ASD, including disruptions in excitatory/inhibitory balance, which are believed to underpin many behavioral features of the disorder.
Therapeutic Approaches Targeting Mitochondrial Dysfunction
Understanding mitochondrial involvement in ASD opens the door to potential targeted therapies. Several interventions are currently being explored:
Antioxidant Therapy: Compounds such as coenzyme Q10, alpha-lipoic acid, and N-acetylcysteine have been investigated for their ability to reduce oxidative stress and improve mitochondrial function.
Mitochondrial Cofactor Supplementation: Nutrients like L-carnitine, B-vitamins, and creatine that support mitochondrial metabolism are being studied for their efficacy in alleviating certain ASD symptoms.
Dietary Strategies: Diets such as the ketogenic diet, which alters energy metabolism to rely more on ketone bodies, have shown potential in improving mitochondrial function and behavior in some individuals with ASD.
While these approaches offer promise, it is essential that treatments are personalized and medically supervised, as mitochondrial involvement varies widely among individuals with ASD.
Conclusion
The emerging link between mitochondrial dysfunction and Autism Spectrum Disorder provides a valuable lens through which to understand this complex condition. By affecting energy production, synaptic regulation, and oxidative balance, mitochondria may play a pivotal role in ASD pathogenesis. Further research is needed to refine our understanding and to develop effective, targeted treatments. Nonetheless, recognizing the role of mitochondria enhances our broader understanding of neurodevelopmental disorders and holds promise for future therapeutic innovations that may improve outcomes for individuals on the autism spectrum.
on as a key biological contributor. Mitochondria are vital organelles responsible for generating cellular energy and maintaining homeostasis, particularly in energy-demanding organs like the brain. Impairments in mitochondrial function can significantly disrupt neural development and have been increasingly observed in individuals with ASD. This article delves into the role of mitochondria in ASD, exploring evidence of mitochondrial abnormalities and their implications for understanding and treating this condition.
Mitochondrial Function and Brain Development
Mitochondria produce adenosine triphosphate (ATP), the energy currency of cells, through a process known as oxidative phosphorylation. This energy production is essential for many brain processes including neurotransmission, synaptic plasticity, and cellular repair. Besides energy generation, mitochondria are also involved in regulating calcium levels, producing reactive oxygen species (ROS), and controlling apoptosis (programmed cell death). All these functions are particularly important during early brain development when neurons are rapidly forming connections and networks.
Mitochondrial Dysfunction in Autism
Abnormal Biochemical Profiles Numerous studies have detected elevated levels of lactate, pyruvate, and alanine in the blood and cerebrospinal fluid of individuals with ASD. These findings suggest a disruption in mitochondrial energy metabolism. Abnormal lactate-to-pyruvate ratios, for example, point to oxidative phosphorylation inefficiencies, implying that mitochondria in ASD-affected individuals are not functioning optimally.
Increased Oxidative Stress Mitochondria are both producers and targets of ROS, and when not properly regulated, ROS can damage DNA, proteins, and lipids. In individuals with ASD, elevated markers of oxidative stress and reduced levels of antioxidants such as glutathione have been reported. This imbalance can contribute to neural inflammation and impair neurodevelopment, possibly exacerbating core ASD symptoms.
Genetic Abnormalities in Mitochondrial DNA Some individuals with ASD exhibit mutations or deletions in mitochondrial DNA (mtDNA). Since mtDNA is crucial for the normal function of the electron transport chain—essential for ATP production—such mutations can compromise cellular energy availability. Furthermore, mitochondrial diseases, which often involve mtDNA mutations, frequently present with neurodevelopmental symptoms overlapping with those of ASD.
Mitochondrial Dynamics and Quality Control Mitochondrial health depends on processes such as fission, fusion, and mitophagy (the removal of damaged mitochondria). In ASD, studies have observed altered expressions of genes involved in these dynamic processes. Imbalances in mitochondrial fission and fusion can lead to dysfunctional mitochondria accumulating in neurons, impairing their function and survival.
Impact on Synaptic Function Efficient synaptic transmission relies heavily on mitochondrial energy. Mitochondria located at synapses help regulate calcium signaling and provide the necessary ATP for neurotransmitter release. Mitochondrial dysfunction may therefore contribute to the synaptic abnormalities frequently observed in ASD, including disruptions in excitatory/inhibitory balance, which are believed to underpin many behavioral features of the disorder.
Therapeutic Approaches Targeting Mitochondrial Dysfunction
Understanding mitochondrial involvement in ASD opens the door to potential targeted therapies. Several interventions are currently being explored:
Antioxidant Therapy: Compounds such as coenzyme Q10, alpha-lipoic acid, and N-acetylcysteine have been investigated for their ability to reduce oxidative stress and improve mitochondrial function.
Mitochondrial Cofactor Supplementation: Nutrients like L-carnitine, B-vitamins, and creatine that support mitochondrial metabolism are being studied for their efficacy in alleviating certain ASD symptoms.
Dietary Strategies: Diets such as the ketogenic diet, which alters energy metabolism to rely more on ketone bodies, have shown potential in improving mitochondrial function and behavior in some individuals with ASD.
While these approaches offer promise, it is essential that treatments are personalized and medically supervised, as mitochondrial involvement varies widely among individuals with ASD.
Conclusion
The emerging link between mitochondrial dysfunction and Autism Spectrum Disorder provides a valuable lens through which to understand this complex condition. By affecting energy production, synaptic regulation, and oxidative balance, mitochondria may play a pivotal role in ASD pathogenesis. Further research is needed to refine our understanding and to develop effective, targeted treatments. Nonetheless, recognizing the role of mitochondria enhances our broader understanding of neurodevelopmental disorders and holds promise for future therapeutic innovations that may improve outcomes for individuals on the autism spectrum.
#Autism Spectrum Disorder#Mitochondrial dysfunction#Mitochondria and autism#Neurodevelopmental disorders#Mitochondrial DNA mutations#Oxidative stress in autism#ATP production#Synaptic dysfunction in ASD#Mitochondrial energy metabolism#Reactive oxygen species (ROS)#Mitochondrial cofactor therapy#Antioxidant therapy for autism#Ketogenic diet and autism#Calcium signaling in neurons#Lactate-to-pyruvate ratio#Autism mitochondrial biomarkers#Mitochondrial fission and fusion#Early brain development#Mitochondrial therapeutic strategies#Neuroinflammation in autism
0 notes
Text
Effect of Pollution on Mitochondria: Mechanisms and Implications for Human Health

Introduction
Rapid industrialization and urban development have significantly increased environmental pollution levels, particularly in urban areas. Air pollution is a complex mixture of gases (like ozone, nitrogen dioxide, carbon monoxide) and particulate matter, with PM2.5 being of particular concern due to its ability to penetrate deeply into the alveoli and even enter the bloodstream.
Mitochondria are critical for ATP synthesis, calcium regulation, and programmed cell death (apoptosis). They also regulate redox signaling and cellular metabolism. Given their central role in maintaining cellular integrity and their sensitivity to oxidative stress, mitochondria are prime targets for damage induced by air pollutants. Mitochondrial dysfunction caused by pollution contributes to a wide range of diseases, including neurodegenerative disorders, cardiovascular diseases, and metabolic syndromes.
Mechanisms of Mitochondrial Damage by Pollution
1. Oxidative Stress and ROS Generation
The most prominent mechanism through which pollution affects mitochondria is oxidative stress. PM2.5 and other pollutants contain transition metals and organic compounds that catalyze the formation of reactive oxygen species (ROS). When ROS production exceeds the cell’s antioxidant capacity, it leads to oxidative damage of mitochondrial lipids, proteins, and DNA.
Pollutant-induced oxidative stress disrupts the electron transport chain (ETC), particularly Complex I and III, which further elevates ROS production. This cycle of ROS-induced ROS release exacerbates mitochondrial damage, leading to a decline in membrane potential and ATP production.
2. Mitochondrial Membrane Potential Disruption
The mitochondrial membrane potential (Δψm) is essential for ATP generation through oxidative phosphorylation. Exposure to air pollutants like diesel exhaust particles and PM2.5 causes depolarization of Δψm. This loss of potential impairs ATP synthesis, alters calcium homeostasis, and activates mitochondrial permeability transition pores (mPTP), promoting cell death.
Electron microscopy studies have shown that pollutant-exposed cells exhibit swollen mitochondria, disrupted cristae, and fragmented networks—hallmarks of severe mitochondrial dysfunction.
3. Mitochondrial DNA (mtDNA) Damage
Unlike nuclear DNA, mtDNA lacks protective histones and has limited repair mechanisms, making it highly vulnerable to ROS. PM2.5 and ozone exposure have been shown to cause strand breaks, deletions, and mutations in mtDNA. This impairs the expression of key mitochondrial proteins, further disrupting the ETC and leading to chronic energy deficits.
Mitochondrial DNA copy number has also been used as a biomarker for oxidative stress in epidemiological studies. Decreased mtDNA content correlates with pollution exposure and poor health outcomes in both children and adults.
4. Induction of Apoptosis and Necrosis
Mitochondria regulate both intrinsic apoptotic and necrotic cell death pathways. Air pollutants trigger apoptosis by promoting the release of pro-apoptotic factors such as cytochrome c, apoptosis-inducing factor (AIF), and Smac/DIABLO into the cytosol. These factors activate caspases and lead to programmed cell death.
Additionally, high levels of ROS and persistent mitochondrial dysfunction can shift the balance toward necrosis—an uncontrolled form of cell death characterized by inflammation and tissue damage.
5. Impaired Mitophagy and Biogenesis
Mitophagy is the selective degradation of damaged mitochondria. Air pollution can inhibit mitophagy by altering signaling pathways involving PINK1 and Parkin, leading to the accumulation of dysfunctional mitochondria. Conversely, some pollutants may overstimulate mitophagy, causing loss of healthy mitochondria.
Furthermore, air pollutants downregulate genes associated with mitochondrial biogenesis, such as PGC-1α, NRF1, and TFAM. This leads to reduced mitochondrial number and impaired cellular resilience to oxidative stress.
Health Implications of Pollution-Induced Mitochondrial Dysfunction
1. Cardiovascular Diseases
Endothelial cells, which line blood vessels, rely on functional mitochondria to regulate vascular tone and integrity. Pollution-induced mitochondrial damage in these cells leads to endothelial dysfunction, reduced nitric oxide production, and increased vascular inflammation—key precursors to atherosclerosis, hypertension, and myocardial infarction.
2. Respiratory Conditions
Inhaled pollutants directly affect lung epithelial and alveolar macrophage mitochondria. Damage to these cells can result in chronic obstructive pulmonary disease (COPD), asthma, and reduced lung function. Mitochondrial dysfunction increases susceptibility to infections and reduces the lung’s ability to clear particulate matter.
3. Neurological Disorders
The brain is particularly sensitive to mitochondrial impairment due to its high energy demand. Pollutants like ultrafine particles can cross the blood-brain barrier and accumulate in neural tissue. Studies show that exposure to PM2.5 induces mitochondrial fragmentation, synaptic dysfunction, and neuroinflammation. These changes are associated with increased risks for Alzheimer’s disease, Parkinson’s disease, and cognitive decline.
4. Metabolic Disorders and Diabetes
Mitochondria are central to metabolic homeostasis. Pollutants disrupt mitochondrial function in adipose tissue, liver, and muscle, leading to insulin resistance and impaired glucose metabolism. Epidemiological studies have linked air pollution exposure to increased incidence of type 2 diabetes and obesity.
5. Reproductive and Developmental Effects
Pollution-induced mitochondrial dysfunction can affect gametogenesis, embryo development, and placental function. Prenatal exposure to air pollution has been associated with low birth weight, preterm birth, and developmental delays—possibly due to mitochondrial damage in placental and fetal tissues.
Therapeutic and Preventive Strategies
1. Antioxidant Supplementation
Antioxidants such as vitamin C, vitamin E, Coenzyme Q10, and N-acetylcysteine (NAC) have shown promise in mitigating ROS-induced mitochondrial damage. Mitochondria-targeted antioxidants like MitoQ and SkQ1 are being explored for their ability to penetrate mitochondrial membranes and neutralize ROS at the source.
2. Lifestyle Interventions
Regular physical activity, a diet rich in antioxidants, and stress management can enhance mitochondrial resilience. Avoiding high-pollution areas and using air purifiers indoors can reduce exposure levels, especially in vulnerable populations.
3. Pharmacological Approaches
New therapies targeting mitochondrial biogenesis, dynamics, and repair mechanisms are under investigation. Drugs modulating PGC-1α activity or enhancing mitophagy may offer therapeutic benefit against pollution-induced mitochondrial dysfunction.
Future Research Directions
More research is needed to:
Clarify dose-response relationships between different pollutants and mitochondrial damage.
Investigate the combined effects of multiple pollutants.
Develop non-invasive biomarkers of mitochondrial dysfunction.
Identify genetic or epigenetic factors that influence individual susceptibility.
Explore targeted therapies to prevent or reverse mitochondrial impairment.
Longitudinal and population-based studies will be key in establishing causal links between pollution, mitochondrial dysfunction, and disease progression.
Conclusion
Mitochondria are critical targets of pollution-induced cellular damage. The mechanisms—including oxidative stress, mtDNA damage, impaired mitophagy, and disrupted bioenergetics—converge to impair cellular function and promote disease. As air pollution levels remain a pressing global concern, understanding and addressing mitochondrial responses to environmental toxins is essential for public health. Preventive measures and therapeutic strategies focused on mitochondrial health could play a crucial role in reducing the disease burden associated with pollution.
#Mitochondrial dysfunction#Air pollution and mitochondria#PM2.5 oxidative stress#Pollution-induced ROS#Mitochondrial membrane potential#Mitochondrial DNA damage#Environmental pollutants and health#Mitochondria and air pollutants#Mitophagy impairment#Reactive oxygen species (ROS)#Fine particulate matter effects#Mitochondrial biogenesis inhibition#Pollution-related metabolic disorders#Mitochondrial-targeted antioxidants#Air pollution neurotoxicity#Mitochondria and cardiovascular health#Pollution and mitochondrial diseases#Mitochondrial response to environmental stress#Airborne particles and cell damage
0 notes
Text
Mitochondrial Dysfunction in Beckers Muscular Dystrophy
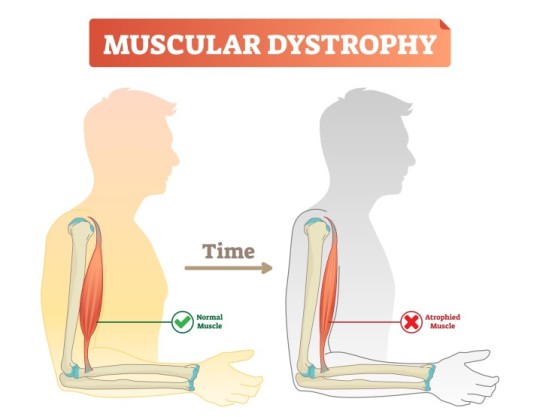
Introduction
Beckers Muscular Dystrophy (BMD) is a genetic neuromuscular disorder caused by mutations in the DMD gene, leading to defective dystrophin production. While dystrophin primarily serves as a structural protein, emerging evidence indicates its role in mitochondrial function and cellular metabolism. This article explores mitochondrial dysfunction in BMD, focusing on bioenergetics, oxidative stress, mitochondrial dynamics, and metabolic consequences.
Bioenergetic Impairment
Mitochondria are the primary energy-producing organelles, generating adenosine triphosphate (ATP) through oxidative phosphorylation (OXPHOS). In BMD, mitochondrial bioenergetics are disrupted due to reduced dystrophin-associated glycoprotein complex (DGC) stability, affecting intracellular signaling and energy metabolism. Studies show that muscle fibers from BMD patients exhibit reduced ATP production, mitochondrial membrane potential (ΔΨm) depolarization, and decreased respiratory chain efficiency. Impaired complex I and complex IV activities have been reported, contributing to decreased oxidative phosphorylation and subsequent muscle weakness.
Oxidative Stress and ROS Accumulation
Mitochondria are a significant source of reactive oxygen species (ROS), which play dual roles as signaling molecules and contributors to oxidative damage. In BMD, excessive ROS production due to dysfunctional electron transport chain (ETC) exacerbates oxidative stress. Studies have demonstrated elevated lipid peroxidation, increased protein carbonylation, and mitochondrial DNA (mtDNA) damage in BMD-affected muscles. Reduced expression of key antioxidant enzymes, such as superoxide dismutase (SOD) and glutathione peroxidase (GPx), further impairs the ability to counteract oxidative damage. The resulting oxidative burden contributes to muscle fiber degeneration, chronic inflammation, and apoptosis.
Mitochondrial Dynamics: Fission and Fusion Imbalance
Mitochondria continuously undergo fission and fusion processes to maintain cellular homeostasis. These dynamics are critical for mitochondrial quality control, ensuring the removal of damaged mitochondria via mitophagy. In BMD, an imbalance between fission and fusion leads to mitochondrial fragmentation and defective turnover. Key regulators such as dynamin-related protein 1 (DRP1) and mitofusin-2 (MFN2) exhibit altered expression, resulting in increased mitochondrial fission and reduced fusion. This dysregulation impairs mitochondrial network integrity, contributing to decreased ATP production and enhanced susceptibility to apoptosis.
Calcium Homeostasis and Mitochondrial Dysfunction
Dystrophin deficiency in BMD disrupts sarcolemmal stability, leading to aberrant calcium (Ca²⁺) handling. Elevated intracellular Ca²⁺ levels induce mitochondrial Ca²⁺ overload, impairing bioenergetic function and promoting mitochondrial permeability transition pore (mPTP) opening. mPTP dysregulation results in mitochondrial swelling, cytochrome c release, and apoptotic cascade activation. Additionally, excessive mitochondrial Ca²⁺ uptake alters ATP synthesis efficiency, exacerbating muscle fiber necrosis and degeneration.
Metabolic Alterations and Energetic Deficits
Skeletal muscle metabolism in BMD is characterized by a shift from oxidative to glycolytic energy production. Defective mitochondrial respiration forces muscle fibers to rely on glycolysis for ATP generation, leading to increased lactate accumulation and metabolic acidosis. This metabolic shift results in early fatigue, reduced endurance, and inefficient energy utilization. Transcriptomic analyses have identified downregulation of genes involved in fatty acid oxidation and tricarboxylic acid (TCA) cycle activity, further confirming the metabolic shift towards glycolysis. Such metabolic alterations compromise muscle function and regeneration capacity, contributing to disease progression.
Mitochondrial Quality Control and Mitophagy Defects
Mitophagy, a selective form of autophagy responsible for degrading damaged mitochondria, is impaired in BMD. The PINK1/Parkin pathway, essential for mitochondrial quality control, is downregulated in dystrophic muscle, leading to the accumulation of dysfunctional mitochondria. Defective mitophagy contributes to mitochondrial swelling, increased oxidative stress, and cellular energy deficits. Additionally, impaired mitophagy reduces the capacity for mitochondrial biogenesis, further exacerbating mitochondrial dysfunction and muscle pathology.
Conclusion
Mitochondrial dysfunction in BMD arises from bioenergetic impairments, oxidative stress, disrupted mitochondrial dynamics, altered Ca²⁺ homeostasis, metabolic deficits, and defective mitophagy. These abnormalities collectively contribute to muscle degeneration and disease progression. Understanding these mitochondrial defects provides valuable insights into the pathophysiology of BMD, emphasizing the need for targeted researc
h to mitigate mitochondrial dysfunction and improve muscle health in affected individuals.
#Beckers Muscular Dystrophy#Mitochondrial dysfunction in BMD#Oxidative stress in muscular dystrophy#Mitochondrial bioenergetics in BMD#ATP production in muscle disease#Reactive oxygen species (ROS) in BMD#Electron transport chain dysfunction#Mitochondrial DNA damage in BMD#Calcium homeostasis in muscular dystrophy#Mitochondrial fission and fusion imbalance#Mitophagy defects in BMD#Muscle fiber degeneration in BMD#Glycolytic metabolism in muscular dystrophy#Mitochondrial membrane potential disruption#Sarcolemmal instability in BMD#Superoxide dismutase (SOD) in muscle health#Mitochondrial permeability transition pore (mPTP)#Fatty acid oxidation in muscle disease#Dystrophin-associated glycoprotein complex (DGC)#Metabolic deficits in Beckers muscular dystrophy
0 notes
Text
Mitochondrial Dysfunction in SLC6A1: A Molecular and Cellular Perspective
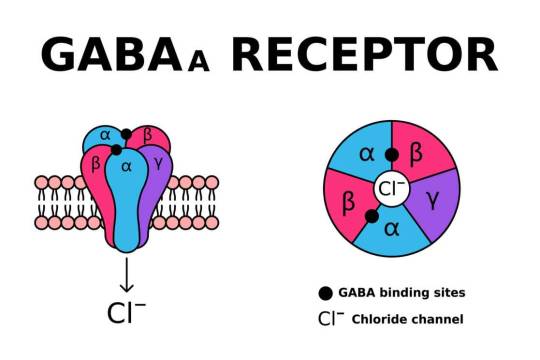
SLC6A1 encodes the gamma-aminobutyric acid (GABA) transporter type 1 (GAT1), a crucial component of inhibitory neurotransmission. Pathogenic variants in SLC6A1 lead to neurological disorders, primarily epilepsy, developmental delay, and neuropsychiatric conditions. While its role in GABAergic signaling is well established, emerging evidence suggests an intersection with mitochondrial dysfunction, which exacerbates disease pathology. This article explores the molecular and cellular mechanisms linking SLC6A1 mutations to mitochondrial impairment, highlighting alterations in energy metabolism, oxidative stress, and mitochondrial dynamics.
1. Introduction The SLC6A1 gene encodes the GAT1 transporter, responsible for reuptaking GABA from the synaptic cleft into presynaptic neurons and astrocytes. Disruptions in SLC6A1 impair inhibitory neurotransmission, contributing to hyperexcitability in neuronal circuits. Recent studies indicate a link between SLC6A1 dysfunction and mitochondrial abnormalities, underscoring a metabolic component to disease pathogenesis. The mitochondrial connection is crucial as these organelles regulate neuronal energy homeostasis and apoptosis. Understanding these mechanisms is essential for dissecting the full scope of SLC6A1-related disorders.
2. Role of SLC6A1 in Cellular and Mitochondrial Function Neurons exhibit high metabolic demand, relying heavily on mitochondria for adenosine triphosphate (ATP) production. GABA metabolism interfaces with mitochondrial pathways, influencing oxidative phosphorylation (OXPHOS) and redox balance. SLC6A1 mutations impair GABA uptake, potentially disrupting mitochondrial function through dysregulated Krebs cycle activity, altered ATP synthesis, and excessive reactive oxygen species (ROS) generation. Additionally, GABAergic dysfunction affects calcium signaling, further impacting mitochondrial integrity.
3. Energy Metabolism and ATP Production Mitochondria generate ATP primarily through OXPHOS. Deficient GABA uptake alters cellular excitability, increasing ATP demand while simultaneously impairing ATP synthesis. Studies show that neurons with SLC6A1 mutations exhibit reduced mitochondrial membrane potential (∆ψm), leading to inefficient ATP generation. Moreover, compensatory glycolysis often fails to meet neuronal energy demands, resulting in cellular stress and neuronal dysfunction.
4. Oxidative Stress and ROS Dysregulation Mitochondria are primary sites of ROS production, which serve as signaling molecules in normal physiology but become deleterious when unregulated. SLC6A1 mutations contribute to ROS imbalance, leading to oxidative stress and lipid peroxidation. Elevated ROS levels have been reported in neurons with impaired GABAergic signaling, suggesting that SLC6A1 mutations exacerbate mitochondrial oxidative damage. This process triggers mitochondrial DNA (mtDNA) mutations, protein oxidation, and lipid peroxidation, further compromising mitochondrial integrity.
5. Calcium Homeostasis and Mitochondrial Dysfunction Neuronal activity depends on tightly regulated calcium homeostasis. Mitochondria buffer intracellular calcium, maintaining synaptic function and preventing excitotoxicity. SLC6A1 dysfunction alters calcium flux due to disrupted GABAergic inhibition, leading to excessive mitochondrial calcium uptake. This triggers the mitochondrial permeability transition pore (mPTP), resulting in bioenergetic failure and apoptotic signaling cascades. Elevated cytosolic calcium further dysregulates mitochondrial enzyme activity, exacerbating metabolic dysfunction.
6. Mitochondrial Dynamics and Biogenesis Mitochondria undergo continuous fission and fusion to adapt to cellular demands. Impaired mitochondrial dynamics are observed in neurons harboring SLC6A1 mutations, leading to fragmented and dysfunctional mitochondria. The fusion-fission imbalance results in defective mitochondrial quality control, accumulation of damaged organelles, and impaired biogenesis. Downregulation of mitophagy-related proteins such as PINK1 and Parkin has been documented in models of SLC6A1 dysfunction, suggesting defective clearance of impaired mitochondria.
7. Synaptic Dysfunction and Mitochondrial Interactions Neurotransmission relies on synaptic mitochondria to meet localized energy demands. GABAergic synapses, in particular, require significant mitochondrial support due to their reliance on ATP-dependent vesicular transport and receptor function. SLC6A1 mutations disrupt synaptic mitochondrial positioning, reducing ATP availability at synapses. This impairment contributes to synaptic dysfunction, decreased inhibitory tone, and aberrant excitatory-inhibitory balance, which are hallmarks of SLC6A1-related neurological disorders.
8. Neuroinflammation and Mitochondrial Dysfunction Mitochondria modulate immune responses through ROS production and inflammatory cytokine signaling. Neurons with SLC6A1 mutations exhibit increased inflammatory markers, such as interleukin-6 (IL-6) and tumor necrosis factor-alpha (TNF-α), indicative of neuroinflammation. Mitochondrial dysfunction exacerbates this process by activating microglia and astrocytes, leading to chronic neuroinflammatory states. This further damages neuronal mitochondria, perpetuating a vicious cycle of dysfunction and degeneration.
9. Genetic and Epigenetic Influences on Mitochondrial Dysfunction Mutations in SLC6A1 not only affect protein function but also influence mitochondrial gene expression and epigenetics. Studies indicate altered expression of nuclear-encoded mitochondrial genes, including those involved in OXPHOS. Additionally, epigenetic modifications such as DNA methylation and histone acetylation impact mitochondrial biogenesis and function in SLC6A1-related disorders. Dysregulated mitochondrial gene transcription exacerbates bioenergetic failure, compounding neurological deficits.
10. Conclusion Mitochondrial dysfunction is an emerging pathological mechanism in SLC6A1-related disorders, contributing to energy deficits, oxidative stress, impaired calcium homeostasis, defective mitochondrial dynamics, and synaptic dysfunction. Understanding the interplay between SLC6A1 mutations and mitochondrial abnormalities provides insights into disease pathogenesis, paving the way for targeted metabolic and neuroprotective interventions. Future research should focus on elucidating the precise molecular pathways linking SLC6A1 dysfunction to mitochondrial pathology, ultimately aiding in the development of novel therapeutic strategies.
#SLC6A1 gene#Mitochondrial dysfunction#GABA transporter (GAT1)#Neurological disorders#Oxidative stress#Mitochondrial energy metabolism#ATP production#Reactive oxygen species (ROS)#Mitochondrial membrane potential#Calcium homeostasis#Neuronal excitability#Mitochondrial biogenesis#Mitochondrial dynamics#Synaptic dysfunction#Neuroinflammation#Mitochondrial quality control#Mitochondrial permeability transition pore (mPTP)#Neurodegeneration#Epigenetic modifications in mitochondria#Mitochondrial oxidative phosphorylation (OXPHOS)
0 notes
Text
Mitochondrial Dysfunction in Primary Mitochondrial Disease
Introduction
Primary Mitochondrial Disease (PMD) refers to a group of genetic disorders resulting from defects in mitochondrial function. Mitochondria play a crucial role in energy production through oxidative phosphorylation (OXPHOS), and their dysfunction leads to a wide spectrum of clinical manifestations affecting multiple organ systems. PMD primarily arises from mutations in mitochondrial DNA (mtDNA) or nuclear DNA (nDNA) encoding mitochondrial proteins, resulting in impaired energy metabolism and increased cellular stress.
Pathophysiology of Mitochondrial Dysfunction
Mitochondrial dysfunction in PMD is primarily caused by defects in the electron transport chain (ETC), which is responsible for ATP synthesis. The ETC comprises five protein complexes embedded in the inner mitochondrial membrane. Mutations affecting these complexes disrupt ATP production, increase the production of reactive oxygen species (ROS), and lead to metabolic imbalances such as lactic acidosis.
Complex I (NADH: ubiquinone oxidoreductase) and Complex IV (cytochrome c oxidase) deficiencies are among the most common defects in PMD. These impairments reduce the efficiency of ATP production, leading to an energy crisis in high-demand tissues such as the brain, muscles, and heart. Additionally, defects in mitochondrial dynamics, including fission and fusion processes, further contribute to cellular dysfunction.
Genetic and Biochemical Basis
PMD is genetically heterogeneous, with mutations in over 350 known genes. These mutations can be inherited in a maternal, autosomal recessive, or dominant manner. Some commonly affected genes include:
MT-ND genes (encoding Complex I subunits)
SURF1 gene (involved in Complex IV assembly)
POLG gene (critical for mtDNA replication and maintenance)
PDHA1 gene (encoding a subunit of the pyruvate dehydrogenase complex)
Mutations in these genes impair the synthesis of key mitochondrial components, leading to energy production failure, oxidative stress, and apoptotic signaling.
Impact on the Nervous System
The nervous system is highly dependent on mitochondrial energy production, making it particularly susceptible to dysfunction. Mitochondrial defects in PMD often manifest as progressive neurodegenerative disorders, including:
Developmental delay and cognitive impairment
Seizures and epilepsy
Hypotonia and muscle weakness
Ataxia and movement disorders
Peripheral neuropathy
Histopathological findings in affected individuals often reveal spongiform degeneration, gliosis, and neuronal loss, particularly in the basal ganglia, cerebellum, and brainstem. These changes contribute to progressive neurological decline.
Effects on Other Organ Systems
Beyond the nervous system, mitochondrial dysfunction in PMD affects multiple organs due to the ubiquitous need for ATP. Key systemic manifestations include:
Musculoskeletal System: Myopathy, exercise intolerance, and rhabdomyolysis are common due to inadequate ATP supply for muscle contraction and maintenance.
Cardiovascular System: Cardiomyopathy, conduction abnormalities, and arrhythmias result from mitochondrial defects in cardiac muscle, leading to impaired contractility and electrical activity.
Metabolic System: Lactic acidosis and metabolic decompensation occur due to defective oxidative metabolism, leading to systemic energy deficits.
Gastrointestinal System: Dysmotility, feeding difficulties, and pancreatic dysfunction are observed, contributing to malnutrition and failure to thrive.
Endocrine System: Mitochondrial dysfunction affects hormone-producing glands, resulting in diabetes, hypothyroidism, and adrenal insufficiency.
Cellular and Molecular Consequences
Mitochondrial dysfunction in PMD leads to several cellular-level consequences, including:
Increased ROS production, causing oxidative stress and damage to lipids, proteins, and DNA.
Dysregulation of apoptosis, leading to premature cell death and tissue degeneration.
Defective calcium homeostasis, impairing neuronal and muscular function.
Impaired mitochondrial biogenesis, reducing the ability of cells to compensate for energy deficits.
Conclusion
Primary Mitochondrial Disease is a complex, multisystem disorder driven by genetic defects in mitochondrial function. The resulting energy production failure impacts the nervous, muscular, cardiovascular, metabolic, and endocrine systems, leading to severe clinical manifestations. Understanding the molecular and biochemical mechanisms underlying PMD is crucial for advancing diagnostic and research efforts. Continued investigation into mitochondrial biology and genetic contributors will enhance our knowledge of this debilitating disease.
#Mitochondrial#Dysfunction#PMD#Energy#Production#System#Defects#Complex#Leading#Mutations#ATP#These#Disease#Genetic#Resulting#Oxidative#Metabolic#Genes#Nervous
0 notes
Text
Mitochondrial Dysfunction in Leigh Syndrome
Introduction:-
Leigh Syndrome (LS) is a rare, severe neurological disorder that typically manifests in infancy or early childhood. It is primarily caused by mitochondrial dysfunction, which results in progressive neurodegeneration. This condition affects approximately 1 in 40,000 newborns and is characterized by lesions in the brainstem and basal ganglia, leading to motor and cognitive impairments.
Pathophysiology of Leigh Syndrome
Mitochondrial dysfunction is central to the pathology of Leigh Syndrome. The mitochondria, often referred to as the powerhouse of the cell, generate adenosine triphosphate (ATP) through oxidative phosphorylation (OXPHOS). This process occurs within the electron transport chain (ETC), which consists of five protein complexes embedded in the inner mitochondrial membrane. In LS, genetic mutations disrupt these complexes, impairing ATP production and causing an accumulation of toxic byproducts such as reactive oxygen species (ROS) and lactate.
The most frequently affected complexes in Leigh Syndrome are Complex I (NADH: ubiquinone oxidoreductase) and Complex IV (cytochrome c oxidase). Mutations in nuclear or mitochondrial DNA (mtDNA) encoding subunits of these complexes lead to decreased enzymatic activity, impairing energy production. As neurons have high energy demands, they are particularly vulnerable to mitochondrial defects, resulting in neuronal cell death and progressive neurodegeneration.
Genetic and Biochemical Basis
Leigh Syndrome is genetically heterogeneous, with over 75 known causative genes. Mutations can be inherited in an autosomal recessive, X-linked, or maternal manner, depending on whether the affected gene is in nuclear DNA or mtDNA. Some of the most common mutations occur in:
MT-ND genes (affecting Complex I)
SURF1 gene (associated with Complex IV deficiency)
PDHA1 gene (disrupting pyruvate dehydrogenase complex, leading to lactic acidosis)
Mitochondrial DNA mutations are maternally inherited, while nuclear DNA mutations follow Mendelian inheritance patterns. The variability in genetic origins contributes to the clinical heterogeneity observed in Leigh Syndrome.
Impact on the Nervous System
Mitochondrial dysfunction in LS predominantly affects the central nervous system (CNS), leading to hallmark neuropathological changes. Bilateral symmetrical lesions appear in the basal ganglia, thalamus, cerebellum, and brainstem. These lesions result from energy deficits and ROS-induced damage, leading to demyelination, gliosis, and neuronal loss.
The neurological symptoms of Leigh Syndrome include:
Developmental delay and regression
Hypotonia (low muscle tone)
Dystonia (involuntary muscle contractions)
Ataxia (lack of muscle coordination)
Ophthalmoplegia (paralysis of eye muscles)
Respiratory failure due to brainstem involvement
As the disease progresses, affected individuals experience worsening motor and cognitive impairments, ultimately leading to severe disability and premature death.
Systemic Effects Beyond the CNS
While Leigh Syndrome primarily affects the nervous system, mitochondrial dysfunction also impacts other organ systems. Metabolic abnormalities such as lactic acidosis arise due to impaired oxidative metabolism, leading to energy deficits in multiple tissues. Additionally, cardiac involvement, such as hypertrophic cardiomyopathy, has been observed in some cases, reflecting the high energy demands of the heart.
The gastrointestinal system may also be affected, with symptoms such as feeding difficulties, failure to thrive, and gastrointestinal dysmotility. This further complicates disease management and contributes to the overall severity of the condition.
Conclusion
Leigh Syndrome is a devastating disorder driven by mitochondrial dysfunction, resulting in widespread neurodegeneration and multi-organ involvement. The genetic heterogeneity and complexity of mitochondrial pathology make it a challenging condition to study and manage. Understanding the molecular basis of mitochondrial dysfunction in LS provides crucial insights into the disease mechanism and potential therapeutic avenues, though treatment remains limited. Continued research into mitochondrial bioenergetics and genetic contributions will be essential in advancing our knowledge of Leigh Syndrome and related mitochondrial disorders.

#Mitochondrial#Leigh#Syndrome#Dysfunction#Leading#Mutations#Genetic#Complex#Energy#LS#Complexes#Affected#DNA#System#Neurodegeneration#Condition#Affects#Lesions#Brainstem
0 notes
Text
Mitochondrial Dysfunction in Spinal Muscular Atrophy (SMA)
Introduction
Spinal Muscular Atrophy (SMA) is a severe neurodegenerative disorder that predominantly affects motor neurons, resulting in progressive muscle weakness and atrophy. The condition is caused by mutations in the survival motor neuron 1 (SMN1) gene, which leads to the loss of SMN protein, a critical factor for motor neuron survival. Although the primary defect lies in the motor neurons, increasing evidence suggests that mitochondrial dysfunction plays a pivotal role in the pathophysiology of SMA. Mitochondria, the powerhouse of the cell, are crucial for cellular energy production and regulation of various metabolic pathways. In the context of SMA, mitochondrial dysfunction has been linked to impaired cellular energy metabolism, oxidative stress, and neuronal death.
This article reviews the emerging role of mitochondrial dysfunction in SMA, examining its impact on motor neurons, the cellular processes involved, and the potential for mitochondrial-targeted therapies.
Mitochondrial Dysfunction in SMA: A Pathophysiological Overview
Mitochondria are essential organelles responsible for generating ATP through oxidative phosphorylation, controlling cellular metabolism, and mediating cell death mechanisms. In SMA, deficits in SMN protein affect multiple cellular pathways, including mitochondrial function. SMN is known to be involved in the biogenesis and maintenance of mitochondria. When its expression is reduced, mitochondrial dysfunction occurs in several ways, contributing to the progressive nature of SMA.
Impaired Mitochondrial Biogenesis
Mitochondrial biogenesis refers to the process by which new mitochondria are formed within cells. This process is tightly regulated by nuclear and mitochondrial signals, with the peroxisome proliferator-activated receptor-gamma coactivator 1-alpha (PGC-1α) being a key regulator of mitochondrial biogenesis. Studies in SMA models have shown that a reduction in SMN protein leads to downregulation of PGC-1α, resulting in decreased mitochondrial biogenesis. This reduced mitochondrial mass is particularly detrimental to motor neurons, which have high energy demands due to their long axonal projections and rapid neurotransmitter signaling.
Mitochondrial Dysfunction and ATP Production
Mitochondrial dysfunction in SMA results in decreased ATP production. ATP is required for essential cellular functions such as protein synthesis, ion transport, and maintaining membrane potential. In motor neurons, impaired ATP generation leads to cellular energy deficits that exacerbate neurodegeneration. Mitochondrial dysfunction also disrupts calcium homeostasis, as mitochondria play a central role in buffering intracellular calcium levels. Elevated intracellular calcium levels can activate enzymes that degrade cellular components, further contributing to cell death in motor neurons.
Oxidative Stress
One of the most significant consequences of mitochondrial dysfunction is the increased production of reactive oxygen species (ROS). Mitochondria are the main source of ROS in cells, and under normal conditions, the antioxidant defense systems neutralize these reactive molecules. However, in SMA, defective mitochondrial function leads to excessive ROS production, which overwhelms the cell’s ability to detoxify them. ROS are highly reactive and can damage cellular structures such as proteins, lipids, and DNA, ultimately contributing to oxidative stress and neuronal injury.
Mitochondrial Dynamics and Morphology
Mitochondrial morphology is highly dynamic, with the organelles undergoing fusion and fission events in response to cellular needs. In SMA, the balance between these processes is disrupted. Studies have shown that reduced SMN levels lead to an increase in mitochondrial fragmentation, a characteristic of mitochondrial dysfunction. Fragmented mitochondria are less efficient in energy production and more prone to damage. Additionally, the fragmented mitochondria in SMA models exhibit impaired mitochondrial transport along axons, further hindering motor neuron function.
Mitochondrial Quality Control
Mitochondrial quality control mechanisms, such as mitophagy, are critical for maintaining mitochondrial health. Mitophagy is the process by which damaged mitochondria are selectively degraded by autophagosomes. In SMA, defects in SMN protein affect the cellular machinery responsible for mitophagy, leading to the accumulation of dysfunctional mitochondria. This impairment in mitochondrial turnover accelerates neurodegeneration by allowing damaged mitochondria to persist, increasing oxidative stress, and triggering cellular apoptosis.
Mitochondrial Dysfunction in Different Types of SMA
SMA is classified into several types based on age of onset and severity, including Type I (Werdnig-Hoffmann disease), Type II, Type III, and Type IV. Mitochondrial dysfunction is observed in all types, but its extent varies depending on the severity of the disease.
SMA Type I
This is the most severe form of SMA, typically presenting in infants before six months of age. These children experience profound muscle weakness and may not survive beyond the first two years of life without intervention. In Type I, mitochondrial dysfunction is particularly pronounced, with severe mitochondrial fragmentation, impaired ATP production, and significant oxidative damage observed in motor neurons. The severity of mitochondrial dysfunction correlates with the extent of neurodegeneration in the spinal cord.
SMA Type II
Type II SMA presents later in infancy or early childhood, with affected individuals showing progressive muscle weakness but with a longer life expectancy compared to Type I. Mitochondrial dysfunction in Type II is still significant but less severe than in Type I. There is evidence of mitochondrial fragmentation and altered mitochondrial dynamics, but motor neurons in Type II patients may still retain some capacity for mitochondrial biogenesis and ATP production, contributing to the slower progression of the disease.
SMA Type III and IV
SMA Type III and IV are milder forms of the disease, with onset typically in childhood or adulthood. While mitochondrial dysfunction is present, it is less pronounced than in Type I and II. In these types, mitochondrial dynamics, ATP production, and oxidative stress are affected, but the clinical presentation is less severe, and individuals often experience a normal or near-normal life expectancy.
Conclusion
Mitochondrial dysfunction is a central feature of the pathophysiology of Spinal Muscular Atrophy (SMA). Reduced SMN protein leads to impaired mitochondrial biogenesis, altered mitochondrial dynamics, increased oxidative stress, and mitochondrial dysfunction. These defects contribute to the progressive degeneration of motor neurons and muscle weakness seen in SMA. Understanding the complex interplay between SMN deficiency and mitochondrial dysfunction provides valuable insights into the disease mechanisms and offers new avenues for therapeutic intervention. Mitochondrial-targeted approaches, including enhancing mitochondrial biogenesis, antioxidant therapy, and modulation of mitochondrial dynamics, hold promise for improving the quality of life and outcomes for SMA patients.
Ongoing research into mitochondrial dysfunction in SMA is crucial for identifying novel treatment strategies that can complement existing therapies and slow disease progression. As therapeutic options evolve, mitochondrial health will likely become an important consideration in the management of SMA, offering hope for more effective treatments in the future.

#Spinal Muscular Atrophy (SMA)#Mitochondrial Dysfunction#Motor Neurons#SMN1 Gene#SMN Protein#Neurodegeneration#Mitochondrial Biogenesis#Oxidative Stress#ATP Production#Mitochondrial Fragmentation#Reactive Oxygen Species (ROS)#Calcium Homeostasis#Mitochondrial Dynamics#Mitochondrial Transport#Mitophagy#Mitochondrial Quality Control#PGC-1α (Peroxisome Proliferator-Activated Receptor-Gamma Coactivator 1-Alpha)#Cellular Energy Metabolism#Mitochondrial-Targeted Therapies#Apoptosis.
0 notes
Text
Mitochondrial Dysfunction in mtARS Disorders
Introduction
Mitochondria are indispensable organelles that facilitate cellular bioenergetics, predominantly through oxidative phosphorylation (OXPHOS). Mitochondrial aminoacyl-tRNA synthetases (mtARS) are essential for the fidelity of mitochondrial translation, catalyzing the ligation of amino acids to their cognate tRNAs. Mutations in mtARS genes precipitate a spectrum of mitochondrial disorders, culminating in dysfunctional protein synthesis and aberrant mitochondrial bioenergetics. This review delves into the molecular pathogenesis of mitochondrial dysfunction in mtARS disorders, elucidating their biochemical perturbations, clinical phenotypes, and emerging therapeutic paradigms.
Molecular Pathophysiology of mtARS Disorders
MtARS enzymes ensure translational accuracy by charging mitochondrial tRNAs with their respective amino acids, a prerequisite for mitochondrial protein biosynthesis. Pathogenic variants in mtARS genes result in defective aminoacylation, perturbing mitochondrial translation and compromising the integrity of the electron transport chain (ETC). These perturbations induce bioenergetic deficits, increased reactive oxygen species (ROS) production, and secondary mitochondrial stress responses, leading to cellular demise.
Genetic Etiology of mtARS Mutations
Dysfunctional mtARS genes such as DARS2, AARS2, RARS2, and YARS2 have been implicated in autosomal recessive mitochondrial disorders. These mutations exhibit tissue-specific phenotypic heterogeneity, with neurological, muscular, and systemic manifestations. For instance, DARS2 mutations drive leukoencephalopathy with brainstem and spinal cord involvement, whereas AARS2 defects result in a constellation of neurodegenerative and ovarian pathologies.
Biochemical and Cellular Consequences
Dysfunctional mtARS enzymes manifest in multifaceted mitochondrial deficits, including impaired translation, defective OXPHOS, and dysregulated mitochondrial proteostasis.
Disruption of Mitochondrial Translation
Impaired aminoacylation abrogates the synthesis of mitochondrially encoded proteins, undermining the assembly of ETC complexes. This translational arrest culminates in defective ATP synthesis and precipitates a systemic energy deficit.
Electron Transport Chain Dysfunction and Bioenergetic Failure
Pathogenic mtARS mutations lead to OXPHOS inefficiencies, reducing mitochondrial membrane potential (Δψm) and ATP output. Perturbed electron flux exacerbates ROS accumulation, instigating oxidative damage and apoptotic cascades.
Mitochondrial Unfolded Protein Response (UPRmt) Activation
Cellular compensatory mechanisms, including UPRmt, are upregulated in response to mitochondrial translation failure. UPRmt mitigates proteotoxic stress via chaperone-mediated protein refolding and degradation pathways. However, chronic UPRmt activation fosters maladaptive stress responses, contributing to progressive cellular degeneration.
Clinical Manifestations
mtARS disorders exhibit phenotypic variability, spanning from mild neuromuscular impairment to severe multisystemic involvement. The pathophysiological hallmark includes disrupted neurological, muscular, and cardiac function.
Neurological Dysfunction
Neurodegeneration is a predominant feature of mtARS disorders, manifesting as ataxia, seizures, intellectual disability, and progressive leukoencephalopathy. Magnetic resonance imaging (MRI) frequently reveals white matter abnormalities, indicative of compromised oligodendrocyte function.
Myopathy and Metabolic Dysregulation
Muscle tissue, with its high ATP demand, is particularly susceptible to mitochondrial dysfunction. Clinical hallmarks include hypotonia, muscle weakness, and exercise intolerance, often concomitant with metabolic anomalies such as lactic acidosis and elevated pyruvate-to-lactate ratios.
Cardiomyopathy and Mitochondrial Energetics
Hypertrophic cardiomyopathy has been observed in YARS2-associated mitochondrial disorders, wherein compromised ATP synthesis in cardiomyocytes disrupts contractile function and electrophysiological stability.
Diagnostic and Functional Evaluation
A combination of genomic, biochemical, and imaging modalities facilitates the diagnosis of mtARS disorders.
Genomic and Transcriptomic Analysis
Whole-exome sequencing (WES) and whole-genome sequencing (WGS) are pivotal for identifying pathogenic mtARS variants. Transcriptomic profiling elucidates perturbations in mitochondrial gene expression networks, further refining diagnostic accuracy.
Functional Mitochondrial Assays
Biochemical assays, including high-resolution respirometry, ATP quantification, and ETC enzymatic profiling, provide insights into mitochondrial bioenergetics. Patient-derived fibroblasts and induced pluripotent stem cells (iPSCs) serve as valuable models for functional interrogation.
Neuroimaging and Biomarker Identification
Advanced imaging modalities such as MR spectroscopy (MRS) detect metabolic derangements, including lactate accumulation in affected brain regions. Circulating mitochondrial-derived peptides and metabolomic signatures are emerging as potential diagnostic biomarkers.
Emerging Therapeutic Strategies
Despite the absence of curative therapies, multiple avenues are under investigation to ameliorate mitochondrial dysfunction in mtARS disorders.
Mitochondria-Directed Antioxidants
Therapeutic compounds such as MitoQ, idebenone, and edaravone aim to attenuate oxidative stress and preserve mitochondrial integrity.
Genetic and RNA-Based Interventions
Gene therapy strategies utilizing adeno-associated virus (AAV)-mediated delivery and CRISPR-based genome editing are being explored for genetic correction of mtARS mutations. Additionally, RNA-based approaches, including antisense oligonucleotides (ASOs) and mRNA replacement therapy, hold promise in restoring mtARS functionality.
Metabolic Modulation and Supportive Therapies
Ketogenic diets, NAD+ precursors (e.g., nicotinamide riboside), and mitochondrial biogenesis activators (e.g., PGC-1α modulators) are under investigation to enhance cellular energy metabolism. Supportive interventions, including physical therapy and neuromuscular rehabilitation, remain integral to patient management.
Conclusion and Future Directions
Mitochondrial dysfunction in mtARS disorders arises from defective mitochondrial translation, OXPHOS perturbation, and maladaptive stress responses. Advances in genomic medicine, mitochondrial therapeutics, and precision medicine approaches are poised to transform the diagnostic and therapeutic landscape. Continued research into mtARS pathobiology, coupled with translational innovations, will be instrumental in developing targeted interventions for affected individuals.

#Mitochondrial dysfunction#Aminoacyl-tRNA synthetases (mtARS)#Oxidative phosphorylation (OXPHOS)#Electron transport chain (ETC)#Reactive oxygen species (ROS)#Mitochondrial translation#Mitochondrial unfolded protein response (UPRmt)#Bioenergetic failure#Neurodegeneration#Leukoencephalopathy#Hypertrophic cardiomyopathy#Myopathy#Whole-exome sequencing (WES)#Whole-genome sequencing (WGS)#ATP synthesis#Gene therapy#CRISPR-based genome editing#RNA-based interventions#Metabolomic biomarkers#Mitochondrial biogenesis
0 notes
Text
Mitochondrial Dysfunction in Type 2 Diabetes
Introduction
Mitochondria, essential for cellular energy metabolism, play a crucial role in bioenergetics and metabolic homeostasis. Mitochondrial dysfunction has been implicated as a key pathophysiological factor in Type 2 Diabetes Mellitus (T2DM), contributing to insulin resistance, metabolic inflexibility, and beta-cell dysfunction. This review explores the intricate mechanisms underlying mitochondrial impairments in T2DM, including defective oxidative phosphorylation, disrupted mitochondrial dynamics, impaired mitophagy, and excessive reactive oxygen species (ROS) generation, with a focus on potential therapeutic interventions targeting mitochondrial pathways.
Mechanistic Insights into Mitochondrial Dysfunction in T2DM
1. Defective Oxidative Phosphorylation and ATP Synthesis
Mitochondrial oxidative phosphorylation (OXPHOS) occurs through the electron transport chain (ETC), comprising Complexes I-IV and ATP synthase (Complex V). In T2DM, evidence suggests a downregulation of mitochondrial ETC activity, particularly in Complex I (NADH:ubiquinone oxidoreductase) and Complex III (cytochrome bc1 complex), leading to reduced ATP synthesis. This dysfunction is often linked to compromised NADH oxidation and inefficient proton gradient formation, resulting in cellular energy deficits and impaired insulin-stimulated glucose uptake.
2. Elevated Reactive Oxygen Species (ROS) and Oxidative Stress
Mitochondria are a primary source of ROS, predominantly generated at Complex I and Complex III during electron leakage. In T2DM, excess substrate influx due to hyperglycemia leads to mitochondrial overactivation, driving excessive ROS production. Elevated ROS induces oxidative damage to mitochondrial DNA (mtDNA), lipids, and proteins, disrupting mitochondrial integrity and function. Oxidative stress further impairs insulin signaling by activating stress-responsive kinases such as c-Jun N-terminal kinase (JNK) and IκB kinase (IKK), contributing to systemic insulin resistance.
3. Mitochondrial Biogenesis and Transcriptional Dysregulation
Mitochondrial biogenesis is regulated by the transcriptional coactivator Peroxisome proliferator-activated receptor-gamma coactivator-1 alpha (PGC-1α), which modulates downstream transcription factors such as Nuclear Respiratory Factors (NRF-1/NRF-2) and Mitochondrial Transcription Factor A (TFAM). In T2DM, PGC-1α expression is downregulated, impairing mitochondrial biogenesis and reducing mitochondrial density, leading to decreased oxidative capacity in metabolically active tissues like skeletal muscle and liver.
4. Disrupted Mitochondrial Dynamics and Mitophagy
Mitochondrial quality control is maintained through dynamic fission and fusion processes. Fission, mediated by Dynamin-related protein 1 (Drp1), is necessary for mitochondrial fragmentation and mitophagy, while fusion, regulated by Mitofusin 1/2 (Mfn1/2) and Optic Atrophy 1 (OPA1), maintains mitochondrial integrity. In T2DM, an imbalance favoring excessive fission leads to mitochondrial fragmentation, impairing energy metabolism and exacerbating insulin resistance. Moreover, defective mitophagy, regulated by PTEN-induced kinase 1 (PINK1) and Parkin, results in the accumulation of dysfunctional mitochondria, further aggravating metabolic dysfunction.
Implications of Mitochondrial Dysfunction in T2DM Pathophysiology
1. Skeletal Muscle Insulin Resistance
Skeletal muscle accounts for ~80% of postprandial glucose uptake, relying on mitochondrial ATP production for insulin-mediated glucose transport. Impaired mitochondrial function in muscle cells reduces oxidative phosphorylation efficiency, promoting a shift towards glycolysis and lipid accumulation, ultimately leading to insulin resistance.
2. Pancreatic Beta-Cell Dysfunction
Mitochondrial ATP production is essential for insulin secretion in pancreatic beta cells. ATP-sensitive potassium channels (K_ATP) regulate glucose-stimulated insulin secretion (GSIS), with ATP/ADP ratios dictating channel closure and depolarization-induced insulin exocytosis. In T2DM, mitochondrial dysfunction leads to inadequate ATP generation, impairing GSIS and reducing insulin secretion capacity. Additionally, oxidative stress-induced beta-cell apoptosis contributes to progressive loss of beta-cell mass.
3. Hepatic Mitochondrial Dysfunction and Lipid Dysregulation
Mitochondrial dysfunction in hepatocytes contributes to hepatic insulin resistance and non-alcoholic fatty liver disease (NAFLD). Impaired fatty acid oxidation due to dysfunctional mitochondria leads to lipid accumulation, exacerbating hepatic insulin resistance and systemic metabolic dysregulation.
Therapeutic Strategies Targeting Mitochondrial Dysfunction
1. Exercise-Induced Mitochondrial Adaptation
Physical activity upregulates PGC-1α expression, enhancing mitochondrial biogenesis and oxidative metabolism. High-intensity interval training (HIIT) and endurance exercise improve mitochondrial efficiency and reduce oxidative stress, mitigating insulin resistance in T2DM patients.
2. Pharmacological Modulation of Mitochondrial Function
Metformin: Enhances mitochondrial complex I activity, reducing hepatic gluconeogenesis and oxidative stress.
Thiazolidinediones (TZDs): Activate PPAR-γ, promoting mitochondrial biogenesis and improving insulin sensitivity.
Mitochondria-targeted Antioxidants: Agents like MitoQ, SkQ1, and SS-31 reduce mitochondrial ROS, preventing oxidative damage and preserving mitochondrial function.
3. Nutritional and Metabolic Interventions
Ketogenic and Low-Carb Diets: Enhance mitochondrial fatty acid oxidation, reducing lipid accumulation and improving metabolic flexibility.
Intermittent Fasting: Induces mitochondrial biogenesis and autophagy, improving metabolic homeostasis.
Nutraceuticals: Coenzyme Q10, resveratrol, and nicotinamide riboside (NR) enhance mitochondrial function and energy metabolism.
4. Emerging Gene and Cellular Therapies
Gene Therapy: Targeted upregulation of PGC-1α and TFAM to restore mitochondrial function.
Mitochondrial Transplantation: Direct transfer of healthy mitochondria to replace dysfunctional ones, an emerging frontier in metabolic disease management.
Conclusion
Mitochondrial dysfunction is a central determinant in the pathogenesis of T2DM, affecting insulin signaling, glucose metabolism, and lipid homeostasis. Targeting mitochondrial pathways through exercise, pharmacological agents, dietary modifications, and emerging gene therapies offers promising avenues for improving metabolic health in T2DM.

#Mitochondrial Dysfunction#Type 2 Diabetes Mellitus (T2DM)#Oxidative Phosphorylation (OXPHOS)#Electron Transport Chain (ETC)#ATP Synthesis#Reactive Oxygen Species (ROS)#Oxidative Stress#Mitochondrial DNA (mtDNA) Damage#Peroxisome Proliferator-Activated Receptor-Gamma Coactivator-1 Alpha (PGC-1α)#Nuclear Respiratory Factors (NRF-1/NRF-2)#Mitochondrial Transcription Factor A (TFAM)#Mitochondrial Biogenesis#Mitochondrial Dynamics (Fission & Fusion)#Dynamin-related protein 1 (Drp1)#Mitofusin 1/2 (Mfn1/2)#Optic Atrophy 1 (OPA1)#Mitophagy#PTEN-Induced Kinase 1 (PINK1)#Parkin#Insulin Resistance#Skeletal Muscle Metabolism#Pancreatic Beta-Cell Dysfunction#Glucose-Stimulated Insulin Secretion (GSIS)#ATP-Sensitive Potassium Channels (K_ATP)#Lipid Dysregulation#Non-Alcoholic Fatty Liver Disease (NAFLD)#Exercise-Induced Mitochondrial Adaptation#High-Intensity Interval Training (HIIT)#Metformin#Thiazolidinediones (TZDs)
0 notes
Text
Mitochondrial Dysfunction in the Pathogenesis of Parkinson’s Disease
Introduction
Parkinson's disease (PD) is a progressive neurodegenerative disorder primarily affecting motor function due to the selective degeneration of dopaminergic neurons in the substantia nigra pars compacta. The pathogenesis of PD is multifactorial, with emerging evidence pointing to mitochondrial dysfunction as a pivotal event in the onset and progression of the disease. This article provides a comprehensive technical analysis of the role of mitochondrial dysfunction in PD, focusing on key molecular mechanisms, genetic factors, and potential therapeutic strategies.
Mitochondria and Their Cellular Roles
Mitochondria are essential organelles that generate the majority of the cell's ATP via oxidative phosphorylation in the electron transport chain (ETC). In addition to their role in energy production, mitochondria are involved in maintaining cellular homeostasis by regulating calcium signaling, apoptosis, and reactive oxygen species (ROS) production. The proper functioning of mitochondria is crucial for neurons, particularly dopaminergic neurons, which have a high metabolic demand.
Mitochondrial Dysfunction and Parkinson's Disease Pathogenesis
Mitochondrial dysfunction in PD primarily manifests through alterations in mitochondrial bioenergetics, increased oxidative stress, defective mitophagy, and calcium dysregulation. These abnormalities converge on exacerbating neuronal injury, particularly in dopaminergic neurons.
1. Impaired Mitochondrial Complex I Activity
One of the hallmark features of mitochondrial dysfunction in PD is the impairment of mitochondrial complex I, the first enzyme complex in the ETC. Complex I is responsible for transferring electrons from NADH to ubiquinone, a critical step in ATP synthesis. Studies consistently show that PD patients exhibit significant reductions in complex I activity in the substantia nigra, which leads to defective ATP production. This mitochondrial dysfunction results in energy deficits, rendering dopaminergic neurons more susceptible to stressors.
Inhibition of complex I activity is not limited to genetic mutations; environmental toxins such as rotenone and paraquat, which inhibit complex I, have been implicated in Parkinsonian syndromes. Furthermore, complex I dysfunction increases the production of ROS, exacerbating oxidative stress in neurons and contributing to mitochondrial damage.
2. Oxidative Stress and ROS Generation
Mitochondria are both the primary source and target of ROS. The process of oxidative phosphorylation inevitably generates ROS as byproducts, particularly superoxide anion, hydrogen peroxide, and hydroxyl radicals. Under normal conditions, ROS are detoxified by endogenous antioxidant systems. However, in PD, mitochondrial dysfunction leads to an imbalance between ROS production and the cell’s antioxidant defenses.
The substantia nigra, which is particularly vulnerable in PD, is exposed to elevated ROS levels due to the high metabolic rate of dopaminergic neurons and the catabolism of dopamine, which generates additional ROS via the action of monoamine oxidase (MAO). Accumulation of ROS results in lipid peroxidation, protein misfolding, and mitochondrial DNA (mtDNA) mutations, all of which contribute to neuronal death and the progression of Parkinson’s pathology.
3. Mitophagy and Dysfunctional Quality Control Mechanisms
Mitophagy, a selective autophagic process that removes damaged or dysfunctional mitochondria, is crucial for maintaining mitochondrial quality and function. In PD, mitophagy is impaired, leading to the accumulation of damaged mitochondria within neurons. The PINK1-parkin pathway plays a pivotal role in the initiation of mitophagy. PINK1, a mitochondrial kinase, accumulates on depolarized mitochondria and recruits the E3 ubiquitin ligase parkin, which ubiquitinates outer mitochondrial membrane proteins to tag them for autophagic degradation.
Mutations in the PINK1 and parkin genes, which are associated with autosomal recessive forms of PD, disrupt this process and contribute to the accumulation of dysfunctional mitochondria. This failure to remove damaged mitochondria exacerbates oxidative stress and promotes the activation of apoptotic signaling pathways. As mitochondrial dysfunction progresses, neuronal survival becomes increasingly compromised, accelerating disease progression.
4. Calcium Homeostasis and Mitochondrial Regulation
Mitochondria play a critical role in buffering cytosolic calcium levels. Neurons, due to their high metabolic activity, are particularly dependent on mitochondrial calcium buffering to prevent cytotoxic calcium overload. However, in PD, mitochondrial dysfunction leads to impaired calcium handling, resulting in an increase in cytosolic calcium concentrations.
Elevated calcium levels activate a variety of calcium-dependent enzymes, such as calpains and phospholipases, that further damage cellular structures. Additionally, excessive calcium in mitochondria can activate the mitochondrial permeability transition pore (mPTP), leading to mitochondrial depolarization, the release of pro-apoptotic factors such as cytochrome c, and eventual cell death.
Genetic Factors in Mitochondrial Dysfunction in PD
Genetic mutations that directly affect mitochondrial function have been identified in familial forms of PD. These mutations often impair mitochondrial dynamics, quality control, and bioenergetics, contributing to the pathogenesis of the disease.
PINK1 and Parkin Mutations: Mutations in the PINK1 gene and the parkin gene, both involved in the regulation of mitophagy, lead to impaired mitochondrial quality control. PINK1, a serine/threonine kinase, normally accumulates on damaged mitochondria and recruits parkin to initiate mitophagy. Loss of PINK1 or parkin function results in the accumulation of dysfunctional mitochondria, contributing to neuronal degeneration.
LRRK2 Mutations: The LRRK2 gene encodes a large protein kinase involved in multiple cellular processes, including mitochondrial dynamics and autophagy. Mutations in LRRK2 are the most common genetic cause of PD, particularly in late-onset forms. LRRK2 is implicated in the regulation of mitochondrial fission and fusion, processes that control mitochondrial morphology and function. Dysregulation of these processes leads to the fragmentation of mitochondria, impaired mitochondrial function, and increased susceptibility to oxidative stress.
Alpha-Synuclein and Mitochondrial Interaction: Alpha-synuclein, the protein most notably associated with Lewy body formation in PD, has also been shown to interact with mitochondrial membranes. Aggregation of alpha-synuclein disrupts mitochondrial dynamics, leading to decreased mitochondrial respiration and increased ROS production. This interaction exacerbates mitochondrial dysfunction and accelerates neurodegeneration.
Environmental Toxins and Mitochondrial Dysfunction
Environmental exposures, particularly to pesticides like rotenone and paraquat, have been shown to inhibit mitochondrial complex I, leading to oxidative stress and mitochondrial dysfunction. These toxins induce PD-like symptoms in animal models, supporting the hypothesis that environmental factors contribute to the pathogenesis of the disease.
Therapeutic Approaches Targeting Mitochondrial Dysfunction
Given the central role of mitochondrial dysfunction in PD, therapeutic strategies aimed at restoring mitochondrial function are being actively explored. These include:
Antioxidant Therapies: Antioxidants such as coenzyme Q10 (CoQ10) have been proposed to alleviate oxidative stress by scavenging ROS. CoQ10 functions as an electron carrier in the ETC and may help restore mitochondrial bioenergetics in PD. Clinical trials, however, have shown mixed results, necessitating further research.
Gene Therapy: Gene therapy approaches aimed at correcting genetic defects that impair mitochondrial function are under investigation. For example, restoring PINK1 or parkin function in neurons may enhance mitophagy and mitigate mitochondrial damage.
Mitochondrial Replacement Therapy: Mitochondrial replacement or mitochondrial transplantation holds promise as a therapeutic strategy for restoring mitochondrial function in PD. Early-stage studies are exploring the feasibility of mitochondrial transplantation into dopaminergic neurons to restore cellular function.
Exercise and Lifestyle Interventions: Regular physical exercise has been shown to stimulate mitochondrial biogenesis and improve mitochondrial function. Exercise-induced upregulation of mitochondrial regulators such as PGC-1α may provide neuroprotective benefits in PD by enhancing mitochondrial turnover and reducing oxidative damage.
Conclusion
Mitochondrial dysfunction is a central event in the pathogenesis of Parkinson's disease, contributing to the degeneration of dopaminergic neurons through mechanisms such as impaired mitochondrial complex I activity, oxidative stress, defective mitophagy, and disrupted calcium homeostasis. Genetic mutations in key mitochondrial regulators such as PINK1, parkin, and LRRK2 exacerbate these defects, while environmental toxins further contribute to mitochondrial damage. Targeting mitochondrial dysfunction through antioxidant therapies, gene therapy, and lifestyle interventions holds promise for mitigating the progression of Parkinson's disease. Understanding the intricate molecular mechanisms linking mitochondrial dysfunction to neurodegeneration in PD will be crucial for developing effective therapeutic strategies.

#Mitochondrial dysfunction#Parkinson’s disease (PD)#Dopaminergic neurons#Substantia nigra#Complex I activity#Oxidative stress#Reactive oxygen species (ROS)#Mitophagy#PINK1#Parkin#Mitochondrial DNA (mtDNA)#Calcium homeostasis#Mitochondrial permeability transition pore(mPTP)#LRRK2#Alpha-synuclein#Mitochondrial dynamics#Genetic mutations in PD#Environmental toxins#Rotenone#Paraquat#Coenzyme Q10 (CoQ10)#Antioxidant therapy#Gene therapy#Mitochondrial replacement therapy#Exercise and mitochondrial biogenesis#Neurodegeneration#Parkinsonian syndromes#Mitochondrial quality control#Mitochondrial fission and fusion#Neuroprotective therapies
0 notes
Text
Mitochondrial Dysfunction in Cardiovascular Disease
Introduction
Mitochondria are essential organelles responsible for the production of cellular energy in the form of adenosine triphosphate (ATP) through oxidative phosphorylation (OXPHOS). The heart, due to its continuous contractile activity, has a high energy demand and is critically dependent on mitochondrial function for normal physiological and pathological processes. Mitochondrial dysfunction has emerged as a central mechanism in the pathogenesis of cardiovascular diseases (CVDs), including ischemic heart disease, heart failure, hypertension, and arrhythmias. This technical overview discusses the molecular mechanisms of mitochondrial dysfunction in cardiovascular disease, its impact on cellular and organ function, and the potential therapeutic strategies to mitigate mitochondrial-related pathophysiology in CVDs.
Mitochondrial Function in Cardiovascular Cells
Mitochondria are highly dynamic organelles that perform several key functions crucial for the health of cardiovascular cells. They are involved in:
ATP Production via Oxidative Phosphorylation: In the mitochondria, energy production is driven by the electron transport chain (ETC), which is composed of complex I-IV embedded in the inner mitochondrial membrane. Electrons derived from NADH and FADH2 produced during the citric acid cycle are transferred through these complexes to ultimately reduce oxygen to water at complex IV. This electron transfer drives proton pumps that create an electrochemical gradient (proton motive force) across the inner mitochondrial membrane, which is utilized by ATP synthase (complex V) to produce ATP.
Calcium Homeostasis: Mitochondria play a crucial role in buffering intracellular calcium concentrations. They take up calcium from the cytoplasm in response to cellular signaling and help maintain cellular homeostasis by storing calcium in the matrix and releasing it when required for cellular signaling. Dysregulation of mitochondrial calcium handling can lead to pathophysiological conditions such as mitochondrial permeability transition (MPT) and cell death.
Reactive Oxygen Species (ROS) Production: Mitochondria are the primary source of ROS due to the incomplete reduction of oxygen molecules during electron transport in the ETC. Under normal conditions, low levels of ROS act as signaling molecules. However, excessive ROS generation due to mitochondrial dysfunction can cause oxidative stress, which damages cellular components such as lipids, proteins, and mitochondrial DNA (mtDNA), contributing to the pathogenesis of cardiovascular diseases.
Apoptosis and Cell Death: Mitochondria are central regulators of apoptosis. The release of pro-apoptotic factors such as cytochrome c from the mitochondrial intermembrane space into the cytoplasm triggers caspase activation, leading to programmed cell death. Mitochondrial dysfunction in cardiovascular tissues can lead to inappropriate cell death, contributing to the progression of CVDs.
Molecular Mechanisms of Mitochondrial Dysfunction in Cardiovascular Disease
Mitochondrial dysfunction in cardiovascular disease can result from several factors, including oxidative damage, altered mitochondrial dynamics, mutations in mitochondrial DNA, and defects in mitochondrial signaling. Below are the primary molecular mechanisms contributing to mitochondrial dysfunction in cardiovascular pathologies:
1. Oxidative Stress and ROS Accumulation
Excessive ROS generation is a hallmark of mitochondrial dysfunction and a major contributor to cardiovascular disease progression. Under normal conditions, the ETC produces ROS as a byproduct of electron transfer; however, under pathological conditions such as ischemia, hypoxia, or heart failure, there is an increase in mitochondrial ROS production. This increase is due to the altered electron flow through the ETC, particularly at complex I and III, which results in the incomplete reduction of oxygen.
The accumulation of ROS causes oxidative damage to mitochondrial lipids, proteins, and mtDNA. For instance, lipid peroxidation of mitochondrial membranes leads to membrane destabilization and disruption of mitochondrial function. ROS also modify proteins involved in mitochondrial dynamics and bioenergetics, impairing the capacity of mitochondria to generate ATP. Furthermore, oxidative damage to mtDNA leads to mutations that compromise the mitochondrial respiratory chain complexes, creating a vicious cycle of mitochondrial dysfunction.
2. Mitochondrial Permeability Transition (MPT) and Calcium Overload
Mitochondrial permeability transition is a critical event in mitochondrial dysfunction. The opening of the mitochondrial permeability transition pore (mPTP) occurs when the inner mitochondrial membrane becomes permeable to ions and small molecules, disrupting the electrochemical gradient required for ATP production. Under pathological conditions such as ischemia-reperfusion injury, excessive ROS and calcium overload activate the mPTP, leading to mitochondrial swelling, loss of membrane potential, and the release of pro-apoptotic factors (e.g., cytochrome c), triggering cell death.
Calcium overload plays a significant role in mitochondrial dysfunction. During stress conditions like ischemia, excessive intracellular calcium is taken up by mitochondria, causing mitochondrial matrix expansion and rupture of the mitochondrial membrane. This exacerbates cellular injury and promotes cell death pathways in the myocardium, contributing to myocardial infarction and heart failure.
3. Mitochondrial Dynamics Dysregulation
Mitochondrial dynamics refer to the continuous processes of mitochondrial fusion and fission that maintain mitochondrial quality and function. In response to cellular stress, mitochondria can undergo fission to segregate damaged components or fusion to promote functional compensation. Mitochondrial fission is regulated by dynamin-related protein 1 (DRP1), while fusion is mediated by mitofusins (MFN1 and MFN2) and optic atrophy 1 (OPA1). In cardiovascular diseases, this dynamic balance is often disrupted, leading to mitochondrial fragmentation, reduced mitochondrial function, and increased susceptibility to apoptosis.
In heart failure, for example, the upregulation of DRP1 and downregulation of fusion proteins contribute to mitochondrial fragmentation, reduced ATP production, and elevated ROS levels. This dysfunction is exacerbated by altered signaling pathways, including those associated with autophagy (mitophagy), which is responsible for removing damaged mitochondria. Dysfunctional mitophagy further impairs mitochondrial quality control, worsening cardiac injury.
4. Mitochondrial DNA Mutations
Mitochondrial DNA is more prone to mutations than nuclear DNA due to its proximity to the ETC and lack of protective histones. In cardiovascular diseases, mutations in mtDNA contribute to defective mitochondrial function. For example, mutations in genes encoding subunits of the OXPHOS complexes (such as ATP6, ND1, or CYTB) lead to impaired ATP synthesis and defective mitochondrial bioenergetics, contributing to myocardial ischemia and heart failure.
Mitochondrial mutations may also affect the regulation of ROS production and the activation of apoptotic pathways, accelerating tissue damage and organ dysfunction.
Therapeutic Approaches Targeting Mitochondrial Dysfunction in Cardiovascular Disease
Given the critical role of mitochondria in cardiovascular disease, several therapeutic strategies have been developed to target mitochondrial dysfunction and restore normal mitochondrial function. These include:
1. Mitochondrial Antioxidants
Mitochondrial-targeted antioxidants, such as MitoQ, MitoTEMPO, and SkQ1, have been developed to specifically target ROS within mitochondria. These compounds aim to reduce oxidative stress, limit mitochondrial damage, and improve mitochondrial function. Clinical studies are ongoing to assess the efficacy of these antioxidants in reducing myocardial injury and improving outcomes in heart failure and ischemic heart disease.
2. Mitochondrial Biogenesis Activation
Stimulating mitochondrial biogenesis to increase the number of functional mitochondria is another potential therapeutic strategy. Activators of peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), a key regulator of mitochondrial biogenesis, are being investigated as potential treatments for heart failure. Exercise training is a natural way to activate PGC-1α and increase mitochondrial function, which has been shown to improve cardiac outcomes in patients with heart failure.
3. MPTP Inhibition
Inhibitors of the mPTP, such as cyclosporine A, have been studied for their potential to prevent ischemia-reperfusion injury by inhibiting pore opening. By preserving mitochondrial integrity, these inhibitors may help reduce myocardial damage and improve survival after myocardial infarction.
4. Gene Therapy and Mitochondrial Transplantation
Gene therapy approaches, including the use of CRISPR/Cas9 to correct mitochondrial DNA mutations, hold promise in treating mitochondrial diseases. Additionally, mitochondrial transplantation, where healthy mitochondria are delivered to damaged cardiac cells, is an emerging area of research, with the potential to restore mitochondrial function and improve heart function in patients with severe myocardial injury.
Conclusion
Mitochondrial dysfunction plays a central role in the pathogenesis of cardiovascular diseases, contributing to impaired ATP production, increased ROS production, and cell death. Understanding the molecular mechanisms underlying mitochondrial dysfunction provides critical insights into the development of novel therapeutic strategies. Approaches targeting mitochondrial biogenesis, oxidative stress, mitochondrial dynamics, and mPTP inhibition offer promising avenues for the treatment of cardiovascular diseases and could lead to more effective management of conditions such as heart failure, ischemic heart disease, and hypertension. However, further research and clinical trials are needed to fully elucidate the potential of these therapeutic strategies in improving cardiovascular health.
#Mitochondria#Cardiovascular Disease (CVD)#Mitochondrial Dysfunction#Oxidative Phosphorylation#ATP Production#Reactive Oxygen Species (ROS)#Mitochondrial DNA (mtDNA)#Mitochondrial Permeability Transition (MPT)#Calcium Homeostasis#Heart Failure#Ischemic Heart Disease#Hypertension#Mitochondrial Dynamics#Mitochondrial Fission and Fusion#Mitochondrial Biogenesis#Mitochondrial Antioxidants#Mitochondrial Targeted Antioxidants#MPTP Inhibitors#Gene Therapy#Mitochondrial Transplantation#Electrochemical Gradient#Mitochondrial Fragmentation#Cytochrome C#Cell Death (Apoptosis)#Peroxisome Proliferator-Activated#Receptor Gamma Coactivator 1-alpha (PGC-1α)
0 notes
Text
Mitochondrial Dysfunction and the Pathogenesis of Alzheimer’s Disease
Introduction
Mitochondria are pivotal organelles responsible for adenosine triphosphate (ATP) production via oxidative phosphorylation (OXPHOS), regulation of calcium homeostasis, reactive oxygen species (ROS) generation, and the initiation of apoptotic pathways. The brain’s high energy demands and sensitivity to oxidative stress make mitochondrial functionality essential for neuronal health. Recent findings underscore the critical role of mitochondrial dysfunction in the molecular mechanisms driving Alzheimer’s disease (AD), a progressive neurodegenerative disorder characterized by memory loss and cognitive decline. This article provides an in-depth exploration of how mitochondrial impairment contributes to AD pathogenesis and examines emerging therapeutic approaches.
Alzheimer’s Disease: Pathological Hallmarks
Alzheimer’s disease is the most prevalent form of dementia and presents neuropathological features including extracellular amyloid-beta (Aβ) plaques, intracellular neurofibrillary tangles (NFTs) of hyperphosphorylated tau, synaptic dysfunction, and neuronal death. Despite decades of research, the precise etiology remains complex, involving genetic predispositions, environmental triggers, and metabolic imbalances. Mitochondrial dysfunction emerges as a central mechanism intertwining these multifactorial contributors.
Mitochondrial Functions in Neurons
Neurons depend on mitochondria for sustaining synaptic transmission, neurotransmitter synthesis, and maintaining ionic gradients essential for action potentials. Beyond energy production, mitochondria modulate intracellular calcium dynamics, synaptic plasticity, and apoptotic signaling, all of which are integral to learning and memory. Impaired mitochondrial function disrupts these processes, resulting in neuronal vulnerability and degeneration.
Mechanisms of Mitochondrial Dysfunction in Alzheimer’s Disease
Oxidative Stress and ROS Dysregulation: Mitochondria are the primary source of ROS, which, under physiological conditions, participate in cellular signaling. In AD, impaired OXPHOS elevates ROS production, leading to oxidative damage of mitochondrial DNA (mtDNA), proteins, and lipids. This oxidative stress contributes to synaptic deficits, neuronal death, and exacerbates Aβ aggregation and tau hyperphosphorylation.
Amyloid-Beta-Mediated Mitochondrial Toxicity: Aβ peptides localize within mitochondria, where they interact with the inner mitochondrial membrane and components of the electron transport chain (ETC). This interaction inhibits ATP synthesis, augments ROS generation, and compromises mitochondrial membrane potential. Additionally, Aβ disrupts axonal transport of mitochondria, depriving synaptic regions of necessary energy supply.
Tau Pathology and Mitochondrial Dysfunction: Hyperphosphorylated tau impairs microtubule stability, disrupting the intracellular trafficking of mitochondria and other organelles. Dysregulated tau also affects mitochondrial dynamics, including fission and fusion processes, leading to an accumulation of damaged and fragmented mitochondria.
Calcium Homeostasis Impairment: Mitochondria act as buffers for cytosolic calcium. In AD, dysregulated calcium signaling exacerbates mitochondrial calcium overload, triggering permeability transition pore (mPTP) opening and initiating apoptotic cascades. Calcium dysregulation also potentiates Aβ aggregation and tau phosphorylation.
Defects in Mitochondrial Dynamics and Mitophagy: Proper mitochondrial function relies on a balance between fission and fusion processes. In AD, this balance is disrupted, leading to mitochondrial fragmentation and a reduction in mitochondrial network integrity. Impaired mitophagy—the selective autophagic clearance of damaged mitochondria—further exacerbates mitochondrial dysfunction and neuronal degeneration.
Genetic Correlations Between Mitochondrial Dysfunction and Alzheimer’s Disease
APOE-ε4 Allele: The APOE-ε4 variant, a significant genetic risk factor for sporadic AD, has been associated with heightened oxidative stress and impaired mitochondrial efficiency.
Presenilin Mutations: Mutations in PSEN1 and PSEN2, linked to familial AD, disrupt calcium signaling and mitochondrial function, exacerbating cellular stress and neuronal loss.
mtDNA Mutations: Increased somatic mutations and deletions in mtDNA observed in AD patients suggest a direct mitochondrial genomic contribution to the disease.
Therapeutic Strategies Targeting Mitochondrial Dysfunction
Antioxidant Therapies:
Mitochondria-targeted antioxidants such as coenzyme Q10, MitoQ, and SS-31 aim to mitigate oxidative stress and preserve mitochondrial integrity.
Enhancement of Mitochondrial Biogenesis:
Pharmacological agents activating PGC-1α (peroxisome proliferator-activated receptor gamma coactivator-1α) enhance mitochondrial biogenesis, improving neuronal energy metabolism.
Calcium Modulation:
Drugs like memantine, an NMDA receptor antagonist, help restore calcium homeostasis, reducing excitotoxicity and mitochondrial stress.
Promotion of Mitophagy:
Urolithin A and other mitophagy-enhancing compounds facilitate the clearance of defective mitochondria, preventing their accumulation and associated toxicity.
Gene-Based Therapies:
Gene-editing technologies such as CRISPR/Cas9 offer potential for correcting mtDNA mutations and modulating genes implicated in mitochondrial quality control.
Lifestyle Interventions:
Dietary Approaches: Ketogenic diets and caloric restriction enhance mitochondrial efficiency and reduce ROS.
Physical Exercise: Regular aerobic activity stimulates mitochondrial biogenesis and improves oxidative resilience.
Optimized Sleep: Adequate sleep promotes mitochondrial repair and the removal of toxic protein aggregates such as Aβ.
Advancements and Research Directions
Emerging research employs cutting-edge technologies like single-cell transcriptomics, super-resolution microscopy, and metabolomics to unravel the mitochondrial mechanisms underlying AD. Novel drug delivery systems targeting mitochondria and the development of nanotechnologies further hold promise for precision therapeutics.
Conclusion
Mitochondrial dysfunction is a cornerstone in the complex pathogenesis of Alzheimer’s disease, intersecting with oxidative stress, Aβ and tau pathology, calcium dysregulation, and impaired dynamics. Targeting mitochondrial pathways through pharmacological interventions, gene therapy, and lifestyle modifications offers a promising avenue for mitigating disease progression. Continued research into mitochondrial biology and its interplay with neurodegeneration is essential for developing transformative therapies for Alzheimer’s disease.

#Mitochondrial dysfunction#Alzheimer’s disease#Neurodegeneration#Oxidative stress#Reactive oxygen species (ROS)#Amyloid-beta (Aβ)#Tau pathology#Calcium homeostasis#Mitochondrial dynamics#Mitophagy#Electron transport chain (ETC)#Mitochondrial biogenesis#ATP production#Apoptosis#APOE-ε4 allele#Presenilin mutations#mtDNA mutations#Synaptic dysfunction#Neurofibrillary tangles (NFTs)#Antioxidant therapies#Gene-based therapies#Lifestyle interventions#Ketogenic diets#Caloric restriction#Physical exercise#Precision therapeutics
0 notes
Text
Mitochondrial Dysfunction Drives Cognitive Decline
Introduction
Mitochondria, often referred to as the powerhouses of the cell, are crucial organelles responsible for energy production through adenosine triphosphate (ATP) synthesis. Beyond their well-known role in energy metabolism, mitochondria regulate a wide range of cellular processes, including calcium homeostasis, reactive oxygen species (ROS) generation, and apoptosis. When mitochondria malfunction, the consequences can be far-reaching, especially for energy-intensive organs like the brain. Recent research highlights mitochondrial dysfunction as a central factor in cognitive decline, contributing to neurodegenerative diseases such as Alzheimer’s, Parkinson’s, and Huntington’s disease. This article explores the mechanisms by which mitochondrial dysfunction impacts cognitive function and discusses potential therapeutic strategies.
The Brain's Energy Demands and Mitochondrial Function
The human brain, despite accounting for only about 2% of body weight, consumes approximately 20% of the body’s energy. Neurons, the primary cells of the nervous system, rely heavily on mitochondrial ATP to sustain synaptic activity, ion gradient maintenance, and neurotransmitter synthesis. Efficient mitochondrial function is critical for maintaining neuronal health and connectivity, which are foundational for learning, memory, and other cognitive processes.
Mechanisms of Mitochondrial Dysfunction in Cognitive Decline
Reduced ATP Production: Mitochondria produce ATP through oxidative phosphorylation (OXPHOS) in the electron transport chain (ETC). Damage to ETC components, often caused by genetic mutations or oxidative stress, can reduce ATP production. Energy-starved neurons may fail to maintain synaptic function, leading to cognitive impairments.
Excessive ROS Generation: While ROS are natural byproducts of mitochondrial activity and play roles in cell signaling, excessive ROS can damage mitochondrial DNA (mtDNA), proteins, and lipids. This oxidative damage exacerbates mitochondrial dysfunction, creating a vicious cycle that contributes to neuronal degeneration.
Impaired Calcium Regulation: Mitochondria help buffer intracellular calcium levels, which are critical for neurotransmitter release and synaptic plasticity. Dysfunctional mitochondria may fail to regulate calcium, leading to excitotoxicity—a condition where excessive calcium causes neuronal injury and death.
Mitochondrial Dynamics: Mitochondria constantly undergo fission (division) and fusion (joining) to adapt to cellular demands and maintain their integrity. Imbalances in these processes can result in fragmented or overly fused mitochondria, impairing their function and transport within neurons.
Mitochondrial Transport Defects: Neurons have long axons and dendrites that require efficient transport of mitochondria to regions of high energy demand, such as synaptic terminals. Dysfunction in mitochondrial transport mechanisms can disrupt synaptic activity and contribute to cognitive decline.
Mitochondrial Dysfunction in Neurodegenerative Diseases
Alzheimer’s Disease (AD): Mitochondrial dysfunction is a hallmark of AD. Amyloid-beta plaques and tau tangles, characteristic of AD, have been shown to impair mitochondrial function. Elevated ROS levels and reduced ATP production exacerbate neuronal loss and cognitive decline in AD.
Parkinson’s Disease (PD): PD is associated with mutations in genes like PINK1 and PARKIN, which regulate mitochondrial quality control. Impaired mitophagy—the process of removing damaged mitochondria—leads to their accumulation, contributing to dopaminergic neuron degeneration and motor as well as cognitive deficits.
Huntington’s Disease (HD): In HD, mutant huntingtin protein interferes with mitochondrial dynamics and function, resulting in energy deficits and increased oxidative stress. These mitochondrial abnormalities contribute to the progressive cognitive and motor decline observed in HD patients.
Diagnostic and Therapeutic Approaches
Biomarkers of Mitochondrial Dysfunction: Advances in molecular biology have identified potential biomarkers, such as altered mtDNA levels, ROS, and metabolites associated with mitochondrial pathways. These biomarkers can aid in early diagnosis and monitoring of neurodegenerative diseases.
Pharmacological Interventions:
Antioxidants: Compounds like coenzyme Q10, vitamin E, and MitoQ target mitochondrial ROS, reducing oxidative damage and preserving mitochondrial function.
Mitochondrial Biogenesis Enhancers: Agents like resveratrol and PGC-1α activators promote the production of new mitochondria and improve mitochondrial health.
Calcium Modulators: Drugs that stabilize calcium levels, such as memantine, may protect neurons from excitotoxicity.
Gene Therapy: Gene-editing tools like CRISPR/Cas9 offer potential to correct mtDNA mutations or enhance the expression of genes involved in mitochondrial quality control. For example, boosting PINK1 or PARKIN expression could improve mitophagy in PD.
Lifestyle Interventions:
Dietary Interventions: Ketogenic diets and intermittent fasting have been shown to enhance mitochondrial function by promoting efficient energy utilization and reducing ROS.
Exercise: Regular physical activity stimulates mitochondrial biogenesis and reduces oxidative stress, offering neuroprotective benefits.
Sleep Optimization: Adequate sleep is essential for mitochondrial repair and the clearance of damaged proteins, such as amyloid-beta.
Future Directions in Research
Understanding the interplay between mitochondrial dysfunction and cognitive decline opens new avenues for research and therapy. Emerging technologies, such as single-cell transcriptomics and advanced imaging, allow for detailed exploration of mitochondrial dynamics in neurons. Additionally, the development of mitochondria-targeted drugs and nanotechnologies holds promise for precise therapeutic interventions.
Conclusion
Mitochondrial dysfunction plays a pivotal role in driving cognitive decline and is implicated in the pathogenesis of various neurodegenerative diseases. Addressing mitochondrial health through targeted therapies, lifestyle modifications, and early diagnostic measures offers hope for mitigating cognitive impairments and improving quality of life. As our understanding of mitochondrial biology deepens, so too does the potential for innovative treatments that could transform the landscape of neurodegenerative disease management.
#Mitochondrial dysfunction#Cognitive decline#Neurodegenerative diseases#Alzheimer\u2019s disease (AD)#Parkinson\u2019s disease (PD)#Huntington\u2019s disease (HD)#ATP production#Oxidative phosphorylation (OXPHOS)#Reactive oxygen species (ROS)#Mitochondrial DNA (mtDNA)#Calcium homeostasis#Synaptic activity#Excitotoxicity#Mitochondrial dynamics#Mitochondrial fission and fusion#Mitophagy#Mitochondrial transport#Biomarkers#Antioxidants#Mitochondrial biogenesis#Gene therapy#CRISPR/Cas9#Lifestyle interventions#Ketogenic diet#Exercise#Sleep optimization#Neuronal health#Therapeutic strategies
0 notes
Text
The Impact of High Fructose Corn Syrup on Mitochondrial Function
The Impact of High Fructose Corn Syrup on Mitochondrial Function:
Analysis
High fructose corn syrup (HFCS), a widely used sweetener derived from corn, has become a major component of the modern diet, especially in processed foods and sugary beverages. HFCS is composed of glucose and fructose in varying proportions, with HFCS-55 (55% fructose, 45% glucose) and HFCS-42 (42% fructose, 58% glucose) being the most common formulations. While the impact of HFCS on metabolic health has been widely discussed, recent studies have shown that it can also exert a detrimental effect on mitochondrial function. This technical analysis explores the biochemical mechanisms by which HFCS damages mitochondria, contributing to cellular dysfunction and a range of metabolic diseases.
Mitochondrial Physiology and Biochemical Function
Mitochondria are highly specialized organelles responsible for producing adenosine triphosphate (ATP), the primary energy currency of the cell, through oxidative phosphorylation (OXPHOS). This process occurs in the inner mitochondrial membrane and involves the electron transport chain (ETC) and ATP synthase. The mitochondria are also involved in regulating cellular metabolism, maintaining redox balance, calcium homeostasis, and apoptosis (programmed cell death). Mitochondrial dysfunction, characterized by impaired ATP production, altered mitochondrial dynamics (fusion/fission), and excessive reactive oxygen species (ROS) production, is a key factor in the pathogenesis of many chronic diseases, including obesity, insulin resistance, cardiovascular diseases, and neurodegenerative disorders.
Fructose Metabolism and Its Divergence from Glucose
The metabolism of fructose, particularly in the liver, diverges significantly from that of glucose, and it is this divergence that underpins much of the mitochondrial dysfunction associated with HFCS consumption. Unlike glucose, which is predominantly metabolized via glycolysis and the citric acid cycle (TCA cycle), fructose bypasses the rate-limiting step of glycolysis, catalyzed by phosphofructokinase-1 (PFK-1), and is instead phosphorylated by fructokinase to form fructose-1-phosphate. This rapid metabolism of fructose in the liver can overwhelm metabolic pathways and lead to the accumulation of intermediate metabolites such as dihydroxyacetone phosphate (DHAP) and glyceraldehyde, which can be further converted to fatty acids and triglycerides through de novo lipogenesis (DNL).
Excessive fructose consumption leads to the accumulation of triglycerides, particularly within hepatocytes, which is a hallmark of non-alcoholic fatty liver disease (NAFLD). The lipid accumulation in the liver, in turn, exacerbates mitochondrial dysfunction and oxidative stress, contributing to insulin resistance and a cascade of metabolic disorders.
Mechanisms of Mitochondrial Damage Induced by HFCS
Increased ROS Production
One of the most significant consequences of excess fructose metabolism is the elevated production of reactive oxygen species (ROS). ROS are byproducts of cellular respiration, primarily generated at complexes I and III of the electron transport chain. Under normal conditions, mitochondria have a robust antioxidant defense system, including enzymes like superoxide dismutase (SOD), catalase, and glutathione peroxidase, which help neutralize ROS. However, when cells are exposed to an overload of fructose, the liver mitochondria become overwhelmed, leading to excessive ROS generation.
Fructose metabolism increases the NADPH/NADP+ ratio, enhancing the activity of nicotinamide adenine dinucleotide phosphate (NADPH)-dependent oxidases such as NADPH oxidase (NOX), which further amplifies ROS production. These ROS cause oxidative damage to mitochondrial DNA (mtDNA), lipids in the mitochondrial membranes, and mitochondrial proteins. Such damage impairs mitochondrial function by decreasing mitochondrial membrane potential, disrupting the electron transport chain, and promoting mitochondrial fragmentation. Furthermore, mtDNA is particularly vulnerable to ROS due to its proximity to the electron transport chain and the lack of histone protection, leading to mutations that impair mitochondrial replication and protein synthesis.
Disruption of Mitochondrial Biogenesis
Mitochondrial biogenesis refers to the process by which new mitochondria are synthesized within a cell to meet the energy demands. This process is tightly regulated by several transcription factors, most notably peroxisome proliferator-activated receptor-gamma coactivator 1-alpha (PGC-1α). PGC-1α activates the transcription of nuclear and mitochondrial genes involved in energy metabolism, mitochondrial dynamics, and antioxidant defenses.
Fructose consumption has been shown to inhibit PGC-1α expression in both liver and skeletal muscle cells. Reduced PGC-1α levels lead to impaired mitochondrial biogenesis, which limits the ability of cells to adapt to increased energy demands. This is particularly concerning in tissues with high metabolic demands, such as muscle, heart, and liver, where impaired mitochondrial function can exacerbate energy deficits and lead to insulin resistance, fatty liver disease, and other metabolic disorders.
Mitochondrial Permeability Transition and Apoptosis
Chronic exposure to high levels of fructose can lead to mitochondrial permeability transition (MPT), a process in which the mitochondrial inner membrane becomes permeable to ions and small molecules, disrupting mitochondrial function. MPT is typically induced by excessive ROS production, calcium overload, or changes in the mitochondrial membrane potential. The opening of the mitochondrial permeability transition pore (MPTP) leads to the loss of mitochondrial membrane potential, uncoupling of oxidative phosphorylation, and the release of pro-apoptotic factors such as cytochrome c into the cytoplasm. This, in turn, activates the caspase cascade, promoting apoptosis.
In the context of HFCS-induced mitochondrial dysfunction, increased ROS and altered metabolic intermediates, such as ceramides, may trigger MPT and apoptotic pathways, leading to cell death and tissue damage. In tissues such as the liver and pancreas, this can exacerbate the pathological progression of fatty liver disease and insulin resistance.
Fatty Acid Accumulation and Impaired Beta-Oxidation
Excessive fructose consumption induces de novo lipogenesis (DNL) in the liver, leading to an increase in the synthesis of fatty acids, which are esterified into triglycerides and stored within hepatocytes. This accumulation of lipids can overwhelm the capacity of mitochondria to oxidize these fatty acids via beta-oxidation, leading to mitochondrial dysfunction. The accumulation of lipotoxic intermediates such as ceramides and diacylglycerols further impairs mitochondrial function by inhibiting key enzymes involved in mitochondrial energy production.
Moreover, the excess fatty acids can impair mitochondrial membrane fluidity, reducing the efficiency of oxidative phosphorylation. The lipid-induced mitochondrial dysfunction leads to further oxidative stress, creating a feedback loop that exacerbates the metabolic disturbances caused by high fructose intake.
Clinical Implications of HFCS-Induced Mitochondrial Dysfunction
The long-term consumption of HFCS has profound implications for human health, particularly in the context of metabolic diseases:
Insulin Resistance and Type 2 Diabetes: HFCS-induced mitochondrial dysfunction, particularly in liver and muscle cells, contributes to impaired insulin signaling and glucose homeostasis. As mitochondrial function declines, cells become less responsive to insulin, leading to insulin resistance, a precursor to type 2 diabetes.
Non-Alcoholic Fatty Liver Disease (NAFLD): The accumulation of fat in the liver, driven by increased fructose metabolism, leads to mitochondrial damage and dysfunction, which exacerbates the progression of NAFLD to non-alcoholic steatohepatitis (NASH), a more severe form of liver disease.
Cardiovascular Disease: Mitochondrial dysfunction in cardiomyocytes can impair ATP production, leading to reduced contractile function and the progression of cardiovascular disease. The increased oxidative stress and inflammatory mediators associated with mitochondrial damage also contribute to vascular injury and atherosclerosis.
Neurodegenerative Diseases: Impaired mitochondrial function in neurons, driven by high fructose intake, may contribute to neurodegenerative diseases such as Alzheimer's and Parkinson's disease, as mitochondria play a critical role in maintaining neuronal health.
Conclusion
High fructose corn syrup exerts a significant impact on mitochondrial function through several interconnected mechanisms. These include the increased production of reactive oxygen species (ROS), inhibition of mitochondrial biogenesis, induction of mitochondrial permeability transition, and the accumulation of toxic lipid intermediates. These disruptions in mitochondrial homeostasis contribute to the development of insulin resistance, non-alcoholic fatty liver disease, and other chronic metabolic diseases. Addressing the widespread consumption of HFCS and reducing dietary fructose intake could be crucial in mitigating mitochondrial dysfunction and preventing associated metabolic disease
#High Fructose Corn Syrup (HFCS)#Mitochondrial Function#Mitochondria#Oxidative Phosphorylation#Reactive Oxygen Species (ROS)#Fructose Metabolism#ATP Production#Mitochondrial Biogenesis#PGC-1α#Mitochondrial Dysfunction#Insulin Resistance#Fatty Liver Disease(NAFLD)#Mitochondrial Permeability Transition (MPT)#Apoptosis#Beta-Oxidation#De Novo Lipogenesis (DNL)#Ceramides#Lipotoxicity#Non-Alcoholic Steatohepatitis (NASH)#Type 2 Diabetes#Cardiovascular Disease#Neurodegenerative Diseases#Fatty Acids#Liver Mitochondria#Metabolic Disorders#Fructose-Induced Oxidative Stress#Cellular Metabolism#Mitochondrial Membrane Potential#Mitochondrial DNA (mtDNA)#Lipid Accumulation
0 notes
Text
Mitochondrial Dysfunction in Endometriosis
A Technical Overview of Cellular Mechanisms
Endometriosis, a common gynecological condition affecting approximately 10% of women during their reproductive years, is characterized by the presence of endometrial-like tissue outside the uterine cavity, most frequently in the ovaries, fallopian tubes, and peritoneal cavity. This ectopic tissue leads to a chronic inflammatory environment, pain, and infertility. While the pathophysiology of endometriosis is not fully understood, recent studies have increasingly highlighted mitochondrial dysfunction as a central feature of the disease. This technical article provides a detailed exploration of the role of mitochondria in endometriosis, examining the molecular and cellular mechanisms through which mitochondrial dysfunction contributes to disease progression.
Mitochondrial Function and Metabolism
Mitochondria are dynamic organelles responsible for numerous vital cellular processes, most notably ATP production through oxidative phosphorylation (OXPHOS). ATP is generated within the mitochondrial matrix by the electron transport chain (ETC), which involves the transfer of electrons from NADH and FADH2 to oxygen molecules, ultimately producing ATP. In addition to ATP production, mitochondria are involved in the regulation of calcium signaling, the maintenance of cellular redox balance, apoptosis, and the synthesis of key metabolites, including lipids and steroids. Mitochondria also contain their own genome (mitochondrial DNA or mtDNA), which encodes essential components of the ETC and mitochondrial protein synthesis machinery.
Mitochondria maintain their function through a balance of fusion and fission, processes that help ensure the organelle's shape, distribution, and response to stress. Mitochondrial dysfunction can arise from an imbalance in these processes, as well as from damage to mitochondrial DNA (mtDNA), excessive reactive oxygen species (ROS) production, and impaired bioenergetic functions. In the context of endometriosis, these disruptions have profound implications for cellular homeostasis and tissue function.
Mitochondrial Dysfunction in Endometriosis
In endometriosis, altered mitochondrial function contributes significantly to the disease's pathology. The following mechanisms are central to understanding how mitochondrial dysfunction drives the progression of endometriosis:
1. Altered Metabolic Shifts: The Warburg Effect
A hallmark of cancerous and proliferative cells is a shift in cellular metabolism, often referred to as the Warburg effect, in which cells preferentially utilize glycolysis over oxidative phosphorylation for ATP production, even in the presence of oxygen. This metabolic reprogramming is also observed in endometriotic cells, particularly in ectopic lesions, where cells exhibit increased glycolytic activity. In these lesions, endometrial cells rely less on mitochondrial OXPHOS and instead preferentially use glycolysis for ATP production, generating lactate as a byproduct.
This metabolic shift supports enhanced cell proliferation and survival under suboptimal conditions, characteristic of the hyperplastic nature of endometriosis. Glycolysis is less efficient in terms of ATP production compared to OXPHOS, yet it provides the necessary metabolic intermediates for cell division and biosynthesis. Additionally, the accumulation of lactate in the extracellular space lowers the local pH, which can exacerbate tissue inflammation and create a microenvironment conducive to the growth and persistence of ectopic lesions.
2. Mitochondrial DNA Damage and Instability
Mitochondria are highly susceptible to damage due to their proximity to ROS-producing processes in the electron transport chain. ROS, which are byproducts of cellular respiration, can damage mitochondrial lipids, proteins, and most notably, mitochondrial DNA (mtDNA). Unlike nuclear DNA, mtDNA is not protected by histones, making it particularly vulnerable to oxidative damage. In endometriosis, there is compelling evidence that mtDNA is significantly damaged in ectopic endometrial tissue. Studies have shown mtDNA deletions, mutations, and increased levels of mtDNA fragmentation in these tissues, which suggest a breakdown in the integrity of mitochondrial function.
The damaged mtDNA further exacerbates mitochondrial dysfunction, impairing the ability of mitochondria to generate ATP through OXPHOS. This, in turn, results in an increased reliance on anaerobic glycolysis, fueling the Warburg effect. Furthermore, mtDNA mutations can impair mitochondrial protein synthesis, leading to dysfunctional mitochondrial complexes and altered cellular bioenergetics, perpetuating a cycle of cellular dysfunction in endometriotic lesions.
3. Oxidative Stress and Inflammation
One of the critical roles of mitochondria is the regulation of cellular redox balance. Under normal conditions, mitochondria produce ROS as part of the electron transport chain. However, when mitochondrial function is compromised—whether due to damage, oxidative stress, or metabolic reprogramming—excess ROS are produced, leading to a state of oxidative stress. In endometriosis, ectopic endometrial tissue exhibits elevated levels of ROS, contributing to a persistent inflammatory environment.
Oxidative stress in endometriotic lesions is amplified by mitochondrial dysfunction and is further exacerbated by the Warburg effect, which generates additional ROS during glycolysis. ROS directly activate inflammatory pathways, particularly through the nuclear factor-kappa B (NF-κB) signaling pathway, leading to the production of pro-inflammatory cytokines such as IL-6, IL-1β, and TNF-α. These cytokines perpetuate the inflammatory response, recruiting immune cells to the site of ectopic lesions, which leads to pain, fibrosis, and the development of adhesions.
Moreover, ROS play a critical role in sensitizing nociceptors, contributing to the chronic pain experienced by women with endometriosis. The interplay between oxidative stress and inflammation forms a vicious cycle that fuels the progression of endometriosis and promotes the growth and persistence of ectopic lesions.
4. Impaired Mitochondrial Dynamics: Fragmentation and Dysfunction
Mitochondria undergo constant fusion and fission, processes that regulate mitochondrial morphology, quality control, and function. Fusion allows for the mixing of mitochondrial contents, which can help dilute damaged components, while fission helps eliminate dysfunctional mitochondria through mitophagy. In endometriosis, there is evidence of disrupted mitochondrial dynamics, particularly an increase in mitochondrial fragmentation. Fragmented mitochondria are less efficient at ATP production and more prone to accumulating damaged proteins and lipids, which further impairs mitochondrial function.
The imbalance between mitochondrial fusion and fission in endometriosis is linked to altered expression of key proteins such as mitofusins (MFN1/2) and dynamin-related protein 1 (DRP1). DRP1-mediated mitochondrial fission is upregulated in endometriotic lesions, contributing to the generation of fragmented mitochondria. These fragmented organelles are associated with increased oxidative stress, apoptosis resistance, and enhanced cell proliferation—features that contribute to the pathogenesis of endometriosis.
5. Apoptosis Resistance and Cell Survival
Mitochondria play a pivotal role in regulating apoptosis through the release of pro-apoptotic factors, such as cytochrome c, from the mitochondrial intermembrane space. These factors initiate the caspase cascade, leading to cell death. However, in endometriosis, ectopic endometrial cells exhibit resistance to apoptosis, allowing them to survive and proliferate abnormally.
Mitochondrial dysfunction in endometriosis leads to alterations in key apoptotic proteins, including Bcl-2 family members, which regulate mitochondrial outer membrane permeabilization (MOMP). The overexpression of anti-apoptotic proteins, such as Bcl-2 and Bcl-xL, and the downregulation of pro-apoptotic proteins, such as Bax and Bak, result in the persistence of damaged cells. This resistance to apoptosis allows for the survival of endometriotic lesions in hostile environments, contributing to the chronic nature of the disease and complicating treatment strategies.
Therapeutic Implications: Targeting Mitochondrial Dysfunction
Given the central role of mitochondrial dysfunction in endometriosis, therapeutic approaches targeting mitochondrial function hold promise for improving disease management. Several potential strategies include:
Antioxidant Therapies: Reducing oxidative stress through antioxidants such as N-acetylcysteine (NAC), Coenzyme Q10 (CoQ10), and vitamin E could help restore mitochondrial function and reduce inflammation in endometriotic tissues.
Modulation of Mitochondrial Dynamics: Targeting proteins involved in mitochondrial fusion and fission, such as DRP1 and MFN2, may help restore mitochondrial morphology and improve bioenergetic function in endometriotic lesions.
Inhibition of Glycolysis: Given the shift toward glycolysis in endometriotic cells, inhibiting key glycolytic enzymes, such as hexokinase or lactate dehydrogenase, may help reduce lesion growth and metabolic reprogramming.
Mitochondrial Biogenesis Stimulation: Activators of PGC-1α, a central regulator of mitochondrial biogenesis, could promote the generation of healthy mitochondria and improve overall cellular metabolism in endometriotic tissue.
Conclusion
Mitochondrial dysfunction is a key contributor to the pathogenesis of endometriosis. Alterations in mitochondrial metabolism, oxidative stress, mitochondrial DNA damage, and impaired apoptotic regulation are central to the disease's progression. Understanding the molecular mechanisms underlying mitochondrial dysfunction in endometriosis provides novel insights into potential therapeutic strategies. Targeting mitochondrial function and bioenergetics could lead to more effective treatments for endometriosis, alleviating its symptoms and improving outcomes for affected women.

#Mitochondrial Dysfunction#Endometriosis#Mitochondrial DNA (mtDNA)#Oxidative Stress#Reactive Oxygen Species (ROS)#Energy Metabolism#Glycolysis#Warburg Effect#Mitochondrial Dynamics#Mitochondrial Fusion and Fission#Mitochondrial Fragmentation#ATP Production#Oxidative Phosphorylation (OXPHOS)#Cell Proliferation#Apoptosis Resistance#Inflammation#Chronic Pain#Mitophagy#Bcl-2 Family Proteins#Cytokine Production#NF-κB Pathway#Mitochondrial Biogenesis#PGC-1α#Pro-inflammatory Cytokines#Fibrosis#Endometrial Tissue#Antioxidant Therapy#Mitochondrial Targeted Therapies#Fertility Impairment#Cellular Metabolism
1 note
·
View note