#Dynamin-related protein 1 (Drp1)
Explore tagged Tumblr posts
Visit Tumblr Blog
Explore Tumblr blogs with no restrictions, modern design and the best experience.
Last Seen Tumblr Blogs





Fun Fact
BuzzFeed published a report claiming that Tumblr was utilized as a distribution channel for Russian agents to influence American voting habits during the 2016 presidential election in Feb 2018.
Text
Mitochondrial Dysfunction in Type 2 Diabetes
Introduction
Mitochondria, essential for cellular energy metabolism, play a crucial role in bioenergetics and metabolic homeostasis. Mitochondrial dysfunction has been implicated as a key pathophysiological factor in Type 2 Diabetes Mellitus (T2DM), contributing to insulin resistance, metabolic inflexibility, and beta-cell dysfunction. This review explores the intricate mechanisms underlying mitochondrial impairments in T2DM, including defective oxidative phosphorylation, disrupted mitochondrial dynamics, impaired mitophagy, and excessive reactive oxygen species (ROS) generation, with a focus on potential therapeutic interventions targeting mitochondrial pathways.
Mechanistic Insights into Mitochondrial Dysfunction in T2DM
1. Defective Oxidative Phosphorylation and ATP Synthesis
Mitochondrial oxidative phosphorylation (OXPHOS) occurs through the electron transport chain (ETC), comprising Complexes I-IV and ATP synthase (Complex V). In T2DM, evidence suggests a downregulation of mitochondrial ETC activity, particularly in Complex I (NADH:ubiquinone oxidoreductase) and Complex III (cytochrome bc1 complex), leading to reduced ATP synthesis. This dysfunction is often linked to compromised NADH oxidation and inefficient proton gradient formation, resulting in cellular energy deficits and impaired insulin-stimulated glucose uptake.
2. Elevated Reactive Oxygen Species (ROS) and Oxidative Stress
Mitochondria are a primary source of ROS, predominantly generated at Complex I and Complex III during electron leakage. In T2DM, excess substrate influx due to hyperglycemia leads to mitochondrial overactivation, driving excessive ROS production. Elevated ROS induces oxidative damage to mitochondrial DNA (mtDNA), lipids, and proteins, disrupting mitochondrial integrity and function. Oxidative stress further impairs insulin signaling by activating stress-responsive kinases such as c-Jun N-terminal kinase (JNK) and IκB kinase (IKK), contributing to systemic insulin resistance.
3. Mitochondrial Biogenesis and Transcriptional Dysregulation
Mitochondrial biogenesis is regulated by the transcriptional coactivator Peroxisome proliferator-activated receptor-gamma coactivator-1 alpha (PGC-1α), which modulates downstream transcription factors such as Nuclear Respiratory Factors (NRF-1/NRF-2) and Mitochondrial Transcription Factor A (TFAM). In T2DM, PGC-1α expression is downregulated, impairing mitochondrial biogenesis and reducing mitochondrial density, leading to decreased oxidative capacity in metabolically active tissues like skeletal muscle and liver.
4. Disrupted Mitochondrial Dynamics and Mitophagy
Mitochondrial quality control is maintained through dynamic fission and fusion processes. Fission, mediated by Dynamin-related protein 1 (Drp1), is necessary for mitochondrial fragmentation and mitophagy, while fusion, regulated by Mitofusin 1/2 (Mfn1/2) and Optic Atrophy 1 (OPA1), maintains mitochondrial integrity. In T2DM, an imbalance favoring excessive fission leads to mitochondrial fragmentation, impairing energy metabolism and exacerbating insulin resistance. Moreover, defective mitophagy, regulated by PTEN-induced kinase 1 (PINK1) and Parkin, results in the accumulation of dysfunctional mitochondria, further aggravating metabolic dysfunction.
Implications of Mitochondrial Dysfunction in T2DM Pathophysiology
1. Skeletal Muscle Insulin Resistance
Skeletal muscle accounts for ~80% of postprandial glucose uptake, relying on mitochondrial ATP production for insulin-mediated glucose transport. Impaired mitochondrial function in muscle cells reduces oxidative phosphorylation efficiency, promoting a shift towards glycolysis and lipid accumulation, ultimately leading to insulin resistance.
2. Pancreatic Beta-Cell Dysfunction
Mitochondrial ATP production is essential for insulin secretion in pancreatic beta cells. ATP-sensitive potassium channels (K_ATP) regulate glucose-stimulated insulin secretion (GSIS), with ATP/ADP ratios dictating channel closure and depolarization-induced insulin exocytosis. In T2DM, mitochondrial dysfunction leads to inadequate ATP generation, impairing GSIS and reducing insulin secretion capacity. Additionally, oxidative stress-induced beta-cell apoptosis contributes to progressive loss of beta-cell mass.
3. Hepatic Mitochondrial Dysfunction and Lipid Dysregulation
Mitochondrial dysfunction in hepatocytes contributes to hepatic insulin resistance and non-alcoholic fatty liver disease (NAFLD). Impaired fatty acid oxidation due to dysfunctional mitochondria leads to lipid accumulation, exacerbating hepatic insulin resistance and systemic metabolic dysregulation.
Therapeutic Strategies Targeting Mitochondrial Dysfunction
1. Exercise-Induced Mitochondrial Adaptation
Physical activity upregulates PGC-1α expression, enhancing mitochondrial biogenesis and oxidative metabolism. High-intensity interval training (HIIT) and endurance exercise improve mitochondrial efficiency and reduce oxidative stress, mitigating insulin resistance in T2DM patients.
2. Pharmacological Modulation of Mitochondrial Function
Metformin: Enhances mitochondrial complex I activity, reducing hepatic gluconeogenesis and oxidative stress.
Thiazolidinediones (TZDs): Activate PPAR-γ, promoting mitochondrial biogenesis and improving insulin sensitivity.
Mitochondria-targeted Antioxidants: Agents like MitoQ, SkQ1, and SS-31 reduce mitochondrial ROS, preventing oxidative damage and preserving mitochondrial function.
3. Nutritional and Metabolic Interventions
Ketogenic and Low-Carb Diets: Enhance mitochondrial fatty acid oxidation, reducing lipid accumulation and improving metabolic flexibility.
Intermittent Fasting: Induces mitochondrial biogenesis and autophagy, improving metabolic homeostasis.
Nutraceuticals: Coenzyme Q10, resveratrol, and nicotinamide riboside (NR) enhance mitochondrial function and energy metabolism.
4. Emerging Gene and Cellular Therapies
Gene Therapy: Targeted upregulation of PGC-1α and TFAM to restore mitochondrial function.
Mitochondrial Transplantation: Direct transfer of healthy mitochondria to replace dysfunctional ones, an emerging frontier in metabolic disease management.
Conclusion
Mitochondrial dysfunction is a central determinant in the pathogenesis of T2DM, affecting insulin signaling, glucose metabolism, and lipid homeostasis. Targeting mitochondrial pathways through exercise, pharmacological agents, dietary modifications, and emerging gene therapies offers promising avenues for improving metabolic health in T2DM.

#Mitochondrial Dysfunction#Type 2 Diabetes Mellitus (T2DM)#Oxidative Phosphorylation (OXPHOS)#Electron Transport Chain (ETC)#ATP Synthesis#Reactive Oxygen Species (ROS)#Oxidative Stress#Mitochondrial DNA (mtDNA) Damage#Peroxisome Proliferator-Activated Receptor-Gamma Coactivator-1 Alpha (PGC-1α)#Nuclear Respiratory Factors (NRF-1/NRF-2)#Mitochondrial Transcription Factor A (TFAM)#Mitochondrial Biogenesis#Mitochondrial Dynamics (Fission & Fusion)#Dynamin-related protein 1 (Drp1)#Mitofusin 1/2 (Mfn1/2)#Optic Atrophy 1 (OPA1)#Mitophagy#PTEN-Induced Kinase 1 (PINK1)#Parkin#Insulin Resistance#Skeletal Muscle Metabolism#Pancreatic Beta-Cell Dysfunction#Glucose-Stimulated Insulin Secretion (GSIS)#ATP-Sensitive Potassium Channels (K_ATP)#Lipid Dysregulation#Non-Alcoholic Fatty Liver Disease (NAFLD)#Exercise-Induced Mitochondrial Adaptation#High-Intensity Interval Training (HIIT)#Metformin#Thiazolidinediones (TZDs)
0 notes
Text
Link between Blood Vessel Inflammation and Malfunctioning Cellular Powerhouses
The vast majority of cells in the human body contain tiny power plants known as mitochondria that generate much of the energy cells use for day-to-day activities. Like a dynamic renewable resource, these little power plants are constantly dividing and uniting in processes called fission and fusion. The balance between fission and fusion is critical for health – especially cardiovascular health.
Now, in new research, scientists at the Lewis Katz School of Medicine at Temple University (LKSOM) have uncovered a novel mechanism by which abnormalities in mitochondrial fission in endothelial cells – the cells that line the inner surface of blood vessels – contribute to inflammation and oxidative stress in the cardiovascular system. They further show how the fission-fusion balance can be stabilized to lower inflammation using salicylate, the main active ingredient in everyday pain-relieving drugs like aspirin.
The groundbreaking research was published online May 11 in the journal Hypertension.
“It was already known that in cardiovascular disease the function of endothelial cells and mitochondria are impacted by inflammation, but we were unsure whether there was a link between the two,” explained Satoru Eguchi, MD, PhD, FAHA, Professor of Physiology and Professor in the Cardiovascular Research Center, Sol Sherry Thrombosis Research Center, and Center for Metabolic Disease Research at LKSOM.
In endothelial cells, chronic inflammation causes mitochondria to become smaller and fragmented. This damaging process is mediated by a molecule known as dynamin-related protein 1 (Drp1). Normally, Drp1 plays a helpful role in maintaining fission-fusion balance. When cells are stressed by inflammation, however, it steps up fission activity, resulting in mitochondrial fragmentation.
“How Drp1 acts to increase mitochondrial fragmentation when endothelial cells are inflamed has been unclear,” Dr. Eguchi said. “But we wondered whether it might interact with nuclear factor (NF)-κB, which oversees the regulation of inflammatory processes and is involved in endothelial dysfunction.”
In endothelial cells, Dr. Eguchi and colleagues stimulated inflammatory pathways that produced mitochondrial fragmentation. They then examined the effects of blocking Drp1 activity and expression. These experiments showed that in cells, Drp1 inhibition suppresses mitochondrial fission, NF-κB activation, and inflammation. Reductions in fission and inflammation were also observed in cells following NF-κB inhibition, as well as in follow-up studies in mice genetically engineered to have less Drp1.

The researchers next determined whether the anti-inflammatory drug salicylate could also reduce mitochondrial fragmentation. Salicylate works by blocking the activity of multiple inflammatory molecules, including NF-κB. As anticipated, in mice, treatment with salicylate attenuated inflammation and mitochondrial fragmentation via its effects on NF-κB and downstream pathways.
“Our findings suggest that salicylate may be able to maintain the balance between mitochondrial fission and fusion under inflammatory conditions,” Dr. Eguchi said. “This observation could have real clinical impact, since salicylate is already used in aspirin and related pain-relievers.”
In future work, Dr. Eguchi plans to explore the influence of aging and other factors on Drp1 and mitochondrial fission in endothelial cells.
“Mitochondrial function declines with aging, but we also know that exercise and diet influence this process. How these factors come together mechanistically to impact vascular health is not fully understood,” Dr. Eguchi explained.
Other researchers who contributed to the new study include Steven J. Forrester, Kyle J. Preston, Hannah A. Cooper, Michael J. Boyer, Kathleen M. Escoto, Anthony J. Poltronetti, Katherine J. Elliott, Ryohei Kuroda, Masashi Miyao, Victor Rizzo, and Rosario Scalia at the Cardiovascular Research Center at LKSOM; Hiromi Sesaki in the Department of Cell Biology at Johns Hopkins School of Medicine in Baltimore, Maryland; and Tomoko Akiyama and Yayoi Kimura in the Advanced Medical Research Center at Yokohama City University in Yokohama, Japan.
The research was supported by funding from the National Institutes of Health, the American Heart Association, and Japan Grant-in-Aid for Scientific Research.
source https://scienceblog.com/516254/link-between-blood-vessel-inflammation-and-malfunctioning-cellular-powerhouses/
0 notes
Text
Quercetin improves endothelial insulin sensitivity in obese mice.
PMID: Acta Biochim Biophys Sin (Shanghai). 2019 Dec 13 ;51(12):1250-1257. PMID: 31781748 Abstract Title: Quercetin improves endothelial insulin sensitivity in obese mice by inhibiting Drp1 phosphorylation at serine 616 and mitochondrial fragmentation. Abstract: Studies have shown that endothelial insulin resistance induced by oxidative stress contributes to vascular dysfunction in metabolic disorders. Quercetin, a natural antioxidant, has been recently shown to exert protective effects on endothelial function. However, the effects of quercetin on endothelial insulin resistance and its underlying mechanism are unclear. Here, we found that chronic oral treatment of obese mice with quercetin increased vascular endothelial insulin sensitivity, accompanied by alleviated mitochondrial fragmentation as revealed by confocal imaging. In addition, western blot analysis showed that quercetin treatment suppressed the levels of dynamin-related protein 1 (Drp1) and phosphorylation at serine 616 in endothelial cells of obese mice. Mechanistically, quercetin specifically suppressed Drp1 phosphorylation at serine 616, whereas it showed little effects on the Drp1 level and its phosphorylation at serine 637 in cultured endothelial cells under oxidative stress. Furthermore, our results also showed that quercetin suppressed Drp1 phosphorylation at serine 616 by inhibiting PKCδ as revealed by western blot analysis. Knockdown of PKCδ with siRNA alleviated the protective effects of quercetin on endothelial-mitochondrial dynamics and insulin sensitivity. These results suggest that chronic oral treatment with quercetin exerts endothelial protective effects through inhibition of PKCδ and the resultant mitochondrial fragmentation.
read more
0 notes
Text
Activation of dynamin-related protein 1 - dependent mitochondria fragmentation and suppression of osteosarcoma by cryptotanshinone
Abstract
Background
Discovering how to regulate mitochondrial function to reduce cancer growth holds great potential for future cancer therapy development. Here we explore the effects of cryptotanshinone (CPT), a natural product derived from Salvia miltiorrhiza, on mitochondria of osteosarcoma (OS) both in vitro and in vivo, and further elucidate the underlying molecular mechanisms.
Methods
Cytotoxicity in the CPT treated OS cells was analyzed by flow cytometry, CCK8, TUNEL assay and colony formation assays. Flow cytometric analysis was performed to evaluate the effect of CPT on cell cycle of OS cells. Mitochondrial morphology was examined by staining with the mitochondrial membrane potential -sensitive fluorochrome, MitoTracker Red (CMXRos). Immunoblotting, confocal-immunofluorescence staining, co-immunoprecipitation were used to examine the expression and interaction between CPT-mediated Drp1 and Bax. Finally, the synergistic effect of CPT on OS cells was validated using a mouse xenograft tumor model.
Results
In this study, we found CPT treatment induced S-phase arrest, apoptosis, and mitochondrial fragmentation in OS cells. CPT also effectively activated caspase-dependent apoptosis, which could be blocked by pan-caspase inhibitor Z-VAD-FMK. Moreover, we herein provide evidence that treatment with CPT resulted in mitochondrial fragmentation, which is mediated by dynamin-related protein 1 (Drp1), a key mediator of mitochondrial fission. Pursuing this observation, downregulation of Drp1 via silencing RNA could abrogate the induction of apoptosis and mitochondrial fragmentation induced by CPT. Finally, we demonstrate that CPT induced Drp1, which interacted directly with Bcl-2-associated X protein (Bax), which contributed to driving Bax translocation from the cytosol to the mitochondria.
Conclusions
Our findings offer insight into the crosstalk between mitochondrial fragmentation and inhibition of osteosarcoma cell growth in response to CPT.
http://bit.ly/2B7Irau
0 notes
Text
IJMS, Vol. 18, Pages 1825: Extract of Sheng-Mai-San Ameliorates Myocardial Ischemia-Induced Heart Failure by Modulating Ca2+-Calcineurin-Mediated Drp1 Signaling Pathways
IJMS, Vol. 18, Pages 1825: Extract of Sheng-Mai-San Ameliorates Myocardial Ischemia-Induced Heart Failure by Modulating Ca2+-Calcineurin-Mediated Drp1 Signaling Pathways
International Journal of Molecular Sciences doi: 10.3390/ijms18091825
Authors: Ye Yang Yushan Tian Siyao Hu Suxia Bi Suxia Li Yuanjia Hu Junping Kou Jin Qi Boyang Yu
Sheng-Mai-San (SMS) is a well-known traditional Chinese medicine (TCM) complex prescription used to treat heart failure (HF) and angina in clinic. However, its potential therapeutic mechanisms remain unclear. The present study evaluated the cardioprotection of extract of SMS (ESMS) on myocardial ischemia (MI)-induced HF, and explored the underlying molecular mechanisms. The results demonstrated that ESMS (728.0 mg/kg) significantly attenuated MI injury-induced HF by improving cardiac function and pathological changes, decreasing lactate dehydrogenase (LDH), creatine kinase (CK) activities, and brain natriuretic peptide (BNP) levels; increasing ATPase activity; and reducing intracellular Ca2+ levels in MI-induced HF mice model. It also significantly decreased the apoptotic index. In vitro, ESMS (400 μg/mL) inhibited mitochondrial-dependent myocardial apoptosis by modulating the expression of caspase-3 and the Bcl-2/Bax ratio, and improved mitochondrial function through increasing mitochondrial membrane potential and cellular ATP content. ESMS restored intracellular Ca2+ and downregulated the expression of Calcineurin A (CnA), thus inhibiting phosphorylation of dynamin-related protein 1 (Drp1) at Ser616 and increasing phosphorylation of Drp1 at Ser637 to prevent cardiomyocyte mitochondrial fission. Above-mentioned results demonstrated ESMS suppressed mitochondrial-mediated apoptosis in oxygen glucose deprivation (OGD) injured H9c2 cardiomyocytes. These findings suggested that ESMS attenuated MI-induced HF by regulating Ca2+ homeostasis and suppressing mitochondrial mediated apoptosis through the modulation of Ca2+-calcineurin-mediated Drp1 signaling pathways. Our results provide insight into the mechanism and clinical applications of SMS and suggest a potential therapeutic strategy for HF.
from # All Medicine by Alexandros G. Sfakianakis via alkiviadis.1961 on Inoreader http://ift.tt/2g9DHtx from OtoRhinoLaryngology - Alexandros G. Sfakianakis via Alexandros G.Sfakianakis on Inoreader http://ift.tt/2wDfPW3
0 notes
Text
Brain cell changes could help control blood sugar levels
New Post has been published on https://type2diabetestreatment.net/diabetes-news/brain-cell-changes-could-help-control-blood-sugar-levels/
Brain cell changes could help control blood sugar levels
Related News
Sickle cell trait could affect diabetes diagnosis results in HbA1c test09 February 2017
Diabetes skin taster technology unveiled that monitors blood glucose levels05 January 2017
FDA endorses use of Dexcom G5 CGM for replacing blood glucose tests21 December 2016
Father brought out of hypo by quick-thinking toddler in Manchester23 November 2016
Small changes in a cluster of brain cells could help maintain blood sugar levels in people with diabetes, research has suggested. Scientists from Yale School of Medicine launched the study because they wanted to understand more about brain neurons, which control appetite, and how they can affect blood glucose levels. "We've found that changes in the size of mitochondria - small [organisms] responsible for energy production - in certain cells in the brain, could be key to maintaining the blood sugar within a safe range," said senior author Professor Sabrina Diano. In a study using mice, the researchers found that mitochondria significantly changed size when driven by a protein called dynamin-related protein 1 (DRP1). DRP1 was present in some of the animals and missing in others. "We found that when DRP1 activity in the neurons was missing, these neurons were more sensitive to changes in glucose levels," added Diano. The researchers were surprised to observe that these changes in small subsets of neurons were significant in raising blood glucose levels during a fasting period. This occurred because the neurons activated a so-called counter-regulatory response to hypoglycemia, according to Diano. "The brain senses lower glucose levels and sends signals to peripheral organs such as the liver to increase glucose production," she explained. The findings have added to the researchers' understanding of "how the body keeps blood sugar levels within a safe range when sugar levels drop, like during fasting, or when they spike after a meal". The results could be used to help prevent hypoglycemia in people with diabetes, however, the chances of seeing this trialed in humans in the foreseeable future is very unlikely. The results appear in the journal Cell Metabolism.Type 2 Diabetes Treatment Type 2 Diabetes Diet Diabetes Destroyer Reviews Original Article
0 notes
Text
Peroxisome proliferator-activated receptor-gamma dependent pathway reduces the phosphorylation of dynamin-related protein 1 and ameliorates hippocampal injury induced by global ischemia in rats.
PubMed: Related Articles Peroxisome proliferator-activated receptor-gamma dependent pathway reduces the phosphorylation of dynamin-related protein 1 and ameliorates hippocampal injury induced by global ischemia in rats. J Biomed Sci. 2016 May 12;23(1):44 Authors: Chuang YC, Lin TK, Yang DI, Yang JL, Liou CW, Chen SD Abstract BACKGROUND: Dynamin-related protein 1 (Drp1) is a mitochondrial fission protein that, upon phosphorylation at serine 616 (p-Drp1(Ser616)), plays a pivotal role in neuronal death after ischemia. In the present study, we hypothesized that peroxisome proliferator-activated receptor-gamma (PPARγ)-dependent pathway can reduce the expression of p-Drp1(Ser616) and ameliorate hippocampal injury induced by global ischemia in rats. RESULTS: We found that pretreatment of the rats with Mdivi-1, a selective Drp1 inhibitor, decreased the level of transient global ischemia (TGI)-induced p-Drp1(Ser616) and reduced cellular contents of oxidized proteins, activated caspase-3 expression as well as the extent of DNA fragmentation. Delivery of siRNA against Drp1 attenuated the expression of p-Drp1(Ser616) that was accompanied by alleviation of the TGI-induced protein oxidation, activated caspase-3 expression and DNA fragmentation in hippocampal proteins. Exogenous application of pioglitazone, a PPARγ agonist, reduced the p-Drp1(Ser616) expression, decreased TGI-induced oxidative stress and activated caspase-3 expression, lessened the extents of DNA fragmentation, and diminished the numbers of TUNEL-positive neuronal cells; all of these effects were reversed by GW9662, a PPARγ antagonist. CONCLUSIONS: Our findings thus indicated that inhibition of TGI-induced p-Drp1(Ser616) expression by Drp1 inhibitor and Drp1-siRNA can decrease protein oxidation, activated caspase-3 expression and neuronal damage in the hippocampal CA1 subfield. PPARγ agonist, through PPARγ-dependent mechanism and via decreasing p-Drp1(Ser616) expression, can exert anti-oxidative and anti-apoptotic effects against ischemic neuronal injury. PMID: 27175924 [PubMed - indexed for MEDLINE] http://dlvr.it/NGwHTT
0 notes
Text
Mitochondrial Dysfunction in Beckers Muscular Dystrophy
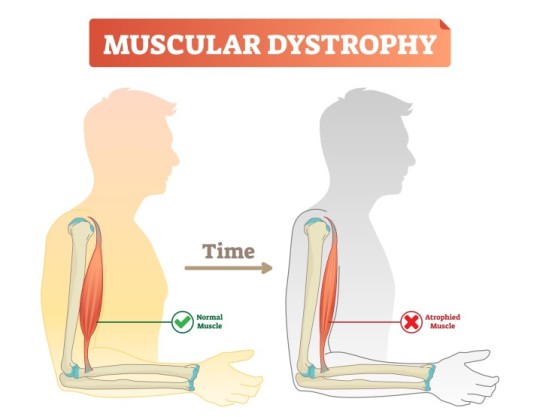
Introduction
Beckers Muscular Dystrophy (BMD) is a genetic neuromuscular disorder caused by mutations in the DMD gene, leading to defective dystrophin production. While dystrophin primarily serves as a structural protein, emerging evidence indicates its role in mitochondrial function and cellular metabolism. This article explores mitochondrial dysfunction in BMD, focusing on bioenergetics, oxidative stress, mitochondrial dynamics, and metabolic consequences.
Bioenergetic Impairment
Mitochondria are the primary energy-producing organelles, generating adenosine triphosphate (ATP) through oxidative phosphorylation (OXPHOS). In BMD, mitochondrial bioenergetics are disrupted due to reduced dystrophin-associated glycoprotein complex (DGC) stability, affecting intracellular signaling and energy metabolism. Studies show that muscle fibers from BMD patients exhibit reduced ATP production, mitochondrial membrane potential (ΔΨm) depolarization, and decreased respiratory chain efficiency. Impaired complex I and complex IV activities have been reported, contributing to decreased oxidative phosphorylation and subsequent muscle weakness.
Oxidative Stress and ROS Accumulation
Mitochondria are a significant source of reactive oxygen species (ROS), which play dual roles as signaling molecules and contributors to oxidative damage. In BMD, excessive ROS production due to dysfunctional electron transport chain (ETC) exacerbates oxidative stress. Studies have demonstrated elevated lipid peroxidation, increased protein carbonylation, and mitochondrial DNA (mtDNA) damage in BMD-affected muscles. Reduced expression of key antioxidant enzymes, such as superoxide dismutase (SOD) and glutathione peroxidase (GPx), further impairs the ability to counteract oxidative damage. The resulting oxidative burden contributes to muscle fiber degeneration, chronic inflammation, and apoptosis.
Mitochondrial Dynamics: Fission and Fusion Imbalance
Mitochondria continuously undergo fission and fusion processes to maintain cellular homeostasis. These dynamics are critical for mitochondrial quality control, ensuring the removal of damaged mitochondria via mitophagy. In BMD, an imbalance between fission and fusion leads to mitochondrial fragmentation and defective turnover. Key regulators such as dynamin-related protein 1 (DRP1) and mitofusin-2 (MFN2) exhibit altered expression, resulting in increased mitochondrial fission and reduced fusion. This dysregulation impairs mitochondrial network integrity, contributing to decreased ATP production and enhanced susceptibility to apoptosis.
Calcium Homeostasis and Mitochondrial Dysfunction
Dystrophin deficiency in BMD disrupts sarcolemmal stability, leading to aberrant calcium (Ca²⁺) handling. Elevated intracellular Ca²⁺ levels induce mitochondrial Ca²⁺ overload, impairing bioenergetic function and promoting mitochondrial permeability transition pore (mPTP) opening. mPTP dysregulation results in mitochondrial swelling, cytochrome c release, and apoptotic cascade activation. Additionally, excessive mitochondrial Ca²⁺ uptake alters ATP synthesis efficiency, exacerbating muscle fiber necrosis and degeneration.
Metabolic Alterations and Energetic Deficits
Skeletal muscle metabolism in BMD is characterized by a shift from oxidative to glycolytic energy production. Defective mitochondrial respiration forces muscle fibers to rely on glycolysis for ATP generation, leading to increased lactate accumulation and metabolic acidosis. This metabolic shift results in early fatigue, reduced endurance, and inefficient energy utilization. Transcriptomic analyses have identified downregulation of genes involved in fatty acid oxidation and tricarboxylic acid (TCA) cycle activity, further confirming the metabolic shift towards glycolysis. Such metabolic alterations compromise muscle function and regeneration capacity, contributing to disease progression.
Mitochondrial Quality Control and Mitophagy Defects
Mitophagy, a selective form of autophagy responsible for degrading damaged mitochondria, is impaired in BMD. The PINK1/Parkin pathway, essential for mitochondrial quality control, is downregulated in dystrophic muscle, leading to the accumulation of dysfunctional mitochondria. Defective mitophagy contributes to mitochondrial swelling, increased oxidative stress, and cellular energy deficits. Additionally, impaired mitophagy reduces the capacity for mitochondrial biogenesis, further exacerbating mitochondrial dysfunction and muscle pathology.
Conclusion
Mitochondrial dysfunction in BMD arises from bioenergetic impairments, oxidative stress, disrupted mitochondrial dynamics, altered Ca²⁺ homeostasis, metabolic deficits, and defective mitophagy. These abnormalities collectively contribute to muscle degeneration and disease progression. Understanding these mitochondrial defects provides valuable insights into the pathophysiology of BMD, emphasizing the need for targeted researc
h to mitigate mitochondrial dysfunction and improve muscle health in affected individuals.
#Beckers Muscular Dystrophy#Mitochondrial dysfunction in BMD#Oxidative stress in muscular dystrophy#Mitochondrial bioenergetics in BMD#ATP production in muscle disease#Reactive oxygen species (ROS) in BMD#Electron transport chain dysfunction#Mitochondrial DNA damage in BMD#Calcium homeostasis in muscular dystrophy#Mitochondrial fission and fusion imbalance#Mitophagy defects in BMD#Muscle fiber degeneration in BMD#Glycolytic metabolism in muscular dystrophy#Mitochondrial membrane potential disruption#Sarcolemmal instability in BMD#Superoxide dismutase (SOD) in muscle health#Mitochondrial permeability transition pore (mPTP)#Fatty acid oxidation in muscle disease#Dystrophin-associated glycoprotein complex (DGC)#Metabolic deficits in Beckers muscular dystrophy
0 notes
Text
Silibinin-induced mitochondria fission leads to mitophagy, which attenuates silibinin-induced apoptosis in MCF-7 and MDA-MB-231 cells.
PMID: Arch Biochem Biophys. 2020 Jan 31:108284. Epub 2020 Jan 31. PMID: 32014401 Abstract Title: Silibinin-induced mitochondria fission leads to mitophagy, which attenuates silibinin-induced apoptosis in MCF-7 and MDA-MB-231 cells. Abstract: We reported previously that higher doses (150-250 μM) of silibinin enhanced fission and inhibited fusion of mitochondria, accompanying apoptosis of double-positive breast cancer cell line MCF-7 cells and triple-negative breast cancer cell line MDA-MB-231 cells. We report here three important questions yet unclarified in the previous study;1) Whether enhanced fission of mitochondria by the treatment of silibinin leads to mitophagy, 2) Whether mitophagy positively contributes to apoptosis and 3) Whether estrogen receptor-positive (ER) MCF-7 cells and estrogen receptor-negative (ER) MDA-MB-231 cells are affected in a different way by silibinin treatment, since silibinin often works through ERs signaling pathway. Mitophagy driven by Pink1/Parkin signaling, plays an important role in eliminating damaged mitochondria. Indeed, increased expression of Pink1 and the recruitment of Parkin andLC3-II to mitochondria by the treatment with silibinin account for silibinin induction of mitophagy. In this study, the effects of mitochondrial division inhibitor 1 (mdivi-1) and small interfering RNA targeting dynamin-related protein 1 (DRP1) were examined to reveal the effect of mitochondrial fission on mitophagy. As expected, mdivi-1 or siRNA targeting DRP1 reversed silibinin-induced mitochondrial fission due to down-regulation in the expression of DRP1. Inhibition of mitochondrial fission by mdivi-1 prevented induction of mitophagy as well as autophagy in both MCF-7 and MDA-MB-231 cells, indicating that silibinin-induced mitochondrial fission leads to mitophagy. Inhibition of mitochondrial fission efficiently prevented silibinin-induced apoptosis in MCF-7 and MDA-MB-231 cells in our previous work, and the second point of the present study, inhibition of mitophagy by Pink1 or Parkin knockdown increased silibinin-induced apoptosis of these cells, respectively, suggesting that the mitophagy induced by silibinin treatment serves as a cytoprotective effect, resulting in reduction of apoptosis of cancer cells in both cells. In the third point, we studied whether estrogen receptors (ERs) played a role in silibinin-induced mitophagy and apoptosis in MCF-7 and MDA-MB-231 cells. ERα and ERβ are not involved in silibinin-induced mitophagic process in MCF-7 and MDA-MB-231 cells. These findings demonstrated that silibinin induced mitochondria fission leads to mitophagy, which attenuates silibinin-induced apoptosis not through ERs-Pink1 or -Parkin pathway in MCF-7 and MDA-MB-231.
read more
0 notes
Text
Silibinin-induced apoptosis of breast cancer cells involves mitochondrial impairment.
PMID: Arch Biochem Biophys. 2019 Aug 15 ;671:42-51. Epub 2019 May 11. PMID: 31085166 Abstract Title: Silibinin-induced apoptosis of breast cancer cells involves mitochondrial impairment. Abstract: Mitochondria are dynamically regulated by fission and fusion processes. Silibinin induces apoptosis of MCF-7 and MDA-MB-231 human breast cancer cells. However, whether or not mitochondria dysfunction is involved in the apoptosis induction with silibinin of both types of the cells remains unknown. We here report that silibinin decreases the mitochondrial mass in terms of MitoTracker Green staining in both breast cancer cells. Silibinin induces morphological changes of mitochondria from oval to truncated or fragmented shapes accordingly. Condensed crests are observed in mitochondria by transmission electron microscopy. Silibinin causes mitochondrial membrane potential reduced. The expression of mitochondrial fission-associated proteins including dynamin-related protein 1 (DRP1) is up-regulated, whereas expression of the mitochondrial fusion-associated proteins, optic atrophy 1 and mitofusin 1, is down-regulated. In addition, silibinin treatment down-regulates ATP content as well as the levels of mitochondrial biogenesis-regulators including mitochondrial transcription factor A, peroxisome proliferator-activated receptor gamma coactivator 1 and nuclear respiratory factor 2. Moreover, treatments with DRP1 inhibitor, mdivi-1, or with DRP1-targetted siRNA efficiently prevent silibinin-induced apoptosis in the breast cancer cells, whereas inhibition of DRP1 phosphorylation with staurosporine increases apoptosis furthermore. Taken together, we conclude that silibinin impairs mitochondrial dynamics and biogenesis, leading to apoptosis of MCF-7 and MDA-MB-123 cells.
read more
0 notes
Text
Silibinin inhibits migration and invasion of breast cancer MDA-MB-231 cells.
PMID: Mol Cell Biochem. 2019 Oct 14. Epub 2019 Oct 14. PMID: 31612353 Abstract Title: Silibinin inhibits migration and invasion of breast cancer MDA-MB-231 cells through induction of mitochondrial fusion. Abstract: Human triple negative breast cancer cells, MDA-MB-231, show typical epithelial to mesenchymal transition associated with cancer progression. Mitochondria play a major role in cancer progression, including metastasis. Changes in mitochondrial architecture affect cellular migration, autophagy and apoptosis. Silibinin is reported to have anti-breast cancer effect. We here report that silibinin at lower concentrations (30-90 μM) inhibits epithelial to mesenchymal transition (EMT) of MDA-MB-231, by increasing the expression of epithelial marker, E-cadherin, and decreasing the expression of mesenchymal markers, N-cadherin and vimentin. Besides, silibinin inhibition of cell migration is associated with reduction in theprotein expression of matrix metalloproteinases 2 and 9 (MMP2 and MMP9) and paxillin. In addition, silibinin treatment increases mitochondrial fusion through down-regulating the expression of mitochondrial fission-associated protein dynamin-related protein 1 (DRP1) and up-regulating the expression of mitochondrial fusion-associated proteins, optic atrophy 1, mitofusin 1 and mitofusin 2. Silibinin perturbed mitochondrial biogenesis via down-regulating the levels of mitochondrial biogenesis regulators including mitochondrial transcription factor A (TFAM), peroxisome proliferator-activated receptor gamma coactivator (PGC1) and nuclear respiratory factor (NRF2). Moreover, DRP1 knockdown or silibinin inhibited cell migration, and MFN1&2 knockdown restored it. Mitochondrial fusion contributes to silibinin's negative effect on cell migration. Silibinin decreased reactive oxygen species (ROS) generation, leading to inhibition of the NLRP3 inflammasome activation. In addition, knockdown of mitofusin 1&2 (MFN 1&2) relieved silibinin-induced inhibition of NLRP3 inflammasome activation. Repression of ROS contributes to the inhibition of the expression of NLRP3, caspase-1 and IL-β proteins as well as of cell migration. Taken together, our study provides evidence that silibinin impairs mitochondrial dynamics and biogenesis, resulting in reduced migration and invasion of the MDA-MB-231 breast cancer cells.
read more
0 notes
Text
Bisphenol A aggravates renal ischemia-reperfusion injury by disrupting mitochondrial homeostasis and N-acetylcysteine mitigates the injurious outcomes.
PMID: IUBMB Life. 2019 Oct 6. Epub 2019 Oct 6. PMID: 31587481 Abstract Title: Bisphenol A aggravates renal ischemia-reperfusion injury by disrupting mitochondrial homeostasis and N-acetylcysteine mitigates the injurious outcomes. Abstract: Exposure to bisphenol A (BPA), a chemical generally used in consumer products, becomes a global public health concern, as humans are increasingly exposed through their daily consuming activities. Renal ischemia-reperfusion (RIR) is the major cause of acute kidney injury with high prevalence and increased long-term risks for multiple comorbidities and mortality. As the kidney is susceptible to these conditions, we explored whether the outcomes following the RIR episode could be influenced by BPA exposure, and investigated the therapeutic possibility by N-acetylcysteine (NAC) including the mechanisms involved. Three groups of male Wistar rats were fed with vehicle, BPA 5, and 50 mg/kg, respectively, for five consecutive weeks then underwent the sham operation. Three other groups with identical treatment underwent bilateral renal IR induction (45-min ischemia followed by 24-hr reperfusion). An additional RIR group was treated with BPA 50 plus NAC 100 mg/kg. BPA-exposedrats that encountered RIR episode showed dose-dependent worsening of RIR injury as evidenced by augmentations of renal dysfunction and histopathological abnormalities, oxidative stress, apoptosis, mitochondrial functional impairment, mitochondrial dynamic, and mitophagy disproportion compared with the vehicle-exposed RIR group. The NAC therapy considerably attenuated the exacerbated effects of BPA, which was associated with increased AMP-activated protein kinase (AMPK), PGC-1α, silent information regulator 3 or sirtuin 3 (SIRT3), and mitofusin 2 (MFN2) expressions but decreased Phosphorylateddynamin-related protein 1 (p-DRP1)/Dynamin-related protein 1 (DRP1), PTEN-induced putative kinase (PINK), and PARKIN expressions. These findings reveal the detrimental effect of repeated BPA exposure on the renal outcomes following the IR episode, and further demonstrate the protective efficacy ofNAC by maintaining mitochondrial homeostasis, which is, partly, mediated through the AMPK-PGC-1α-SIRT3 axis.
read more
0 notes
Text
Inhibiting crosstalk between MET signaling and mitochondrial dynamics and morphology: a novel therapeutic approach for lung cancer and mesothelioma.
Related Articles
Inhibiting crosstalk between MET signaling and mitochondrial dynamics and morphology: a novel therapeutic approach for lung cancer and mesothelioma.
Cancer Biol Ther. 2018 Oct 12;:1-10
Authors: Wang J, Mirzapoiazova T, Carol Tan YH, Pang KM, Pozhitkov A, Wang Y, Wang Y, Mambetsariev B, Wang E, Nasser MW, Batra SK, Raz D, Reckamp K, Kulkarni P, Zheng Y, Salgia R
Abstract The receptor tyrosine kinase MET is frequently involved in malignant transformation and inhibiting its activity in MET-dependent cancers is associated with improved clinical outcomes. Emerging evidence also suggests that mitochondria play an essential role in tumorigenesis and Dynamin Related Protein (DRP1), a key component of the mitochondrial fission machinery, has emerged as an attractive therapeutic target. Here, we report that inhibiting MET activity with the tyrosine kinase inhibitor MGCD516 attenuates viability, migration, and invasion of non-small cell lung cancer (NSCLC) and malignant pleural mesothelioma (MPM) cell lines in vitro, and significantly retards tumor growth in vivo. Interestingly, MGCD516 treatment also results in altered mitochondrial morphology in these cell lines. Furthermore, inhibiting MET pharmacologically or knocking down its expression using siRNA, decreases DRP1 activity alluding to possible crosstalk between them in these two cancers. Consistently, a combination of MGCD516 and mdivi-1, a quinazolinone reported to inhibit mitochondrial fission, is more effective in attenuating proliferation of NSCLC and MPM cell lines than either drug alone. Considered together, the present study has uncovered a novel mechanism underlying mitochondrial regulation by MET that involves crosstalk with DRP1, and suggests that a combination therapy targeting both MET and DRP1 could be a novel strategy for NSCLC and MPM.
PMID: 30311833 [PubMed - as supplied by publisher]
https://ift.tt/2QPFfWI
0 notes
Text
Amplification of glyceronephosphate O-acyltransferase and recruitment of USP30 stabilize DRP1 to promote hepatocarcinogenesis
Hepatocellular carcinoma is a leading cause of cancer-related death worldwide and the underlying pathophysiology of HCC is highly complex. In this study, we report that, in a bioinformatic screen of 2783 genes encoding metabolic enzymes, GNPAT, which encodes the enzyme glyceronephosphate O-acyltransferase, is amplified, upregulated, and highly correlated with poor clinical outcome in human HCC patients. High GNPAT expression in HCC was due to its amplification and transcriptional activation by the c-Myc/KDM1A complex. GNPAT compensated the oncogenic phenotypes in c-Myc-depleted HCC cells. Mechanistically, GNPAT recruited the enzyme USP30, which deubiquitylated and stabilized dynamin-related protein 1 (DRP1), thereby facilitating regulation of mitochondrial morphology, lipid metabolism, and hepatocarcinogenesis. Inhibition of GNPAT and DRP1 dramatically attenuated lipid metabolism and hepatocarcinogenesis. Furthermore, DRP1 mediated the oncogenic phenotypes driven by GNPAT. Taken together, these results indicate that GNPAT and USP30-mediated stabilizaiton of DRP1 play a critical role in the development of HCC. https://ift.tt/2Nhch0D
0 notes
Text
Syntaphilin Ubiquitination Regulates Mitochondrial Dynamics And Tumor Cell Movements
Syntaphilin (SNPH) inhibits the movement of mitochondria in tumor cells, preventing their accumulation at the cortical cytoskeleton and limiting the bioenergetics of cell motility and invasion. Although this may suppress metastasis, the regulation of the SNPH pathway is not well understood. Using a global proteomics screen, we show that SNPH associates with multiple regulators of ubiquitin-dependent responses and is ubiquitinated by the E3 ligase CHIP (or STUB1) on Lys111 and Lys153 in the microtubule-binding domain. SNPH ubiquitination did not result in protein degradation, but instead anchored SNPH on tubulin to inhibit mitochondrial motility and cycles of organelle fusion and fission i.e. dynamics. Expression of ubiquitination-defective SNPH mutant Lys111→Arg or Lys153→Arg increased the speed and distance traveled by mitochondria, repositioned mitochondria to the cortical cytoskeleton, and supported heightened tumor chemotaxis, invasion, and metastasis in vivo. Interference with SNPH ubiquitination activated mitochondrial dynamics, resulting in increased recruitment of the fission regulator dynamin-related protein-1 (Drp1) to mitochondria, and Drp1-dependent tumor cell motility. These data uncover non-degradative ubiquitination of SNPH as a key regulator of mitochondrial trafficking and tumor cell motility and invasion. In this way, SNPH may function as a unique, ubiquitination-regulated suppressor of metastasis https://ift.tt/2JOHyJz
0 notes