#Presenilin mutations
Explore tagged Tumblr posts
Visit Tumblr Blog
Explore Tumblr blogs with no restrictions, modern design and the best experience.
Last Seen Tumblr Blogs





Fun Fact
The total number of visits Tumblr.com received during January 2021 is 327 million.
Text
Mitochondrial Dysfunction and the Pathogenesis of Alzheimer’s Disease
Introduction
Mitochondria are pivotal organelles responsible for adenosine triphosphate (ATP) production via oxidative phosphorylation (OXPHOS), regulation of calcium homeostasis, reactive oxygen species (ROS) generation, and the initiation of apoptotic pathways. The brain’s high energy demands and sensitivity to oxidative stress make mitochondrial functionality essential for neuronal health. Recent findings underscore the critical role of mitochondrial dysfunction in the molecular mechanisms driving Alzheimer’s disease (AD), a progressive neurodegenerative disorder characterized by memory loss and cognitive decline. This article provides an in-depth exploration of how mitochondrial impairment contributes to AD pathogenesis and examines emerging therapeutic approaches.
Alzheimer’s Disease: Pathological Hallmarks
Alzheimer’s disease is the most prevalent form of dementia and presents neuropathological features including extracellular amyloid-beta (Aβ) plaques, intracellular neurofibrillary tangles (NFTs) of hyperphosphorylated tau, synaptic dysfunction, and neuronal death. Despite decades of research, the precise etiology remains complex, involving genetic predispositions, environmental triggers, and metabolic imbalances. Mitochondrial dysfunction emerges as a central mechanism intertwining these multifactorial contributors.
Mitochondrial Functions in Neurons
Neurons depend on mitochondria for sustaining synaptic transmission, neurotransmitter synthesis, and maintaining ionic gradients essential for action potentials. Beyond energy production, mitochondria modulate intracellular calcium dynamics, synaptic plasticity, and apoptotic signaling, all of which are integral to learning and memory. Impaired mitochondrial function disrupts these processes, resulting in neuronal vulnerability and degeneration.
Mechanisms of Mitochondrial Dysfunction in Alzheimer’s Disease
Oxidative Stress and ROS Dysregulation: Mitochondria are the primary source of ROS, which, under physiological conditions, participate in cellular signaling. In AD, impaired OXPHOS elevates ROS production, leading to oxidative damage of mitochondrial DNA (mtDNA), proteins, and lipids. This oxidative stress contributes to synaptic deficits, neuronal death, and exacerbates Aβ aggregation and tau hyperphosphorylation.
Amyloid-Beta-Mediated Mitochondrial Toxicity: Aβ peptides localize within mitochondria, where they interact with the inner mitochondrial membrane and components of the electron transport chain (ETC). This interaction inhibits ATP synthesis, augments ROS generation, and compromises mitochondrial membrane potential. Additionally, Aβ disrupts axonal transport of mitochondria, depriving synaptic regions of necessary energy supply.
Tau Pathology and Mitochondrial Dysfunction: Hyperphosphorylated tau impairs microtubule stability, disrupting the intracellular trafficking of mitochondria and other organelles. Dysregulated tau also affects mitochondrial dynamics, including fission and fusion processes, leading to an accumulation of damaged and fragmented mitochondria.
Calcium Homeostasis Impairment: Mitochondria act as buffers for cytosolic calcium. In AD, dysregulated calcium signaling exacerbates mitochondrial calcium overload, triggering permeability transition pore (mPTP) opening and initiating apoptotic cascades. Calcium dysregulation also potentiates Aβ aggregation and tau phosphorylation.
Defects in Mitochondrial Dynamics and Mitophagy: Proper mitochondrial function relies on a balance between fission and fusion processes. In AD, this balance is disrupted, leading to mitochondrial fragmentation and a reduction in mitochondrial network integrity. Impaired mitophagy—the selective autophagic clearance of damaged mitochondria—further exacerbates mitochondrial dysfunction and neuronal degeneration.
Genetic Correlations Between Mitochondrial Dysfunction and Alzheimer’s Disease
APOE-ε4 Allele: The APOE-ε4 variant, a significant genetic risk factor for sporadic AD, has been associated with heightened oxidative stress and impaired mitochondrial efficiency.
Presenilin Mutations: Mutations in PSEN1 and PSEN2, linked to familial AD, disrupt calcium signaling and mitochondrial function, exacerbating cellular stress and neuronal loss.
mtDNA Mutations: Increased somatic mutations and deletions in mtDNA observed in AD patients suggest a direct mitochondrial genomic contribution to the disease.
Therapeutic Strategies Targeting Mitochondrial Dysfunction
Antioxidant Therapies:
Mitochondria-targeted antioxidants such as coenzyme Q10, MitoQ, and SS-31 aim to mitigate oxidative stress and preserve mitochondrial integrity.
Enhancement of Mitochondrial Biogenesis:
Pharmacological agents activating PGC-1α (peroxisome proliferator-activated receptor gamma coactivator-1α) enhance mitochondrial biogenesis, improving neuronal energy metabolism.
Calcium Modulation:
Drugs like memantine, an NMDA receptor antagonist, help restore calcium homeostasis, reducing excitotoxicity and mitochondrial stress.
Promotion of Mitophagy:
Urolithin A and other mitophagy-enhancing compounds facilitate the clearance of defective mitochondria, preventing their accumulation and associated toxicity.
Gene-Based Therapies:
Gene-editing technologies such as CRISPR/Cas9 offer potential for correcting mtDNA mutations and modulating genes implicated in mitochondrial quality control.
Lifestyle Interventions:
Dietary Approaches: Ketogenic diets and caloric restriction enhance mitochondrial efficiency and reduce ROS.
Physical Exercise: Regular aerobic activity stimulates mitochondrial biogenesis and improves oxidative resilience.
Optimized Sleep: Adequate sleep promotes mitochondrial repair and the removal of toxic protein aggregates such as Aβ.
Advancements and Research Directions
Emerging research employs cutting-edge technologies like single-cell transcriptomics, super-resolution microscopy, and metabolomics to unravel the mitochondrial mechanisms underlying AD. Novel drug delivery systems targeting mitochondria and the development of nanotechnologies further hold promise for precision therapeutics.
Conclusion
Mitochondrial dysfunction is a cornerstone in the complex pathogenesis of Alzheimer’s disease, intersecting with oxidative stress, Aβ and tau pathology, calcium dysregulation, and impaired dynamics. Targeting mitochondrial pathways through pharmacological interventions, gene therapy, and lifestyle modifications offers a promising avenue for mitigating disease progression. Continued research into mitochondrial biology and its interplay with neurodegeneration is essential for developing transformative therapies for Alzheimer’s disease.

#Mitochondrial dysfunction#Alzheimer’s disease#Neurodegeneration#Oxidative stress#Reactive oxygen species (ROS)#Amyloid-beta (Aβ)#Tau pathology#Calcium homeostasis#Mitochondrial dynamics#Mitophagy#Electron transport chain (ETC)#Mitochondrial biogenesis#ATP production#Apoptosis#APOE-ε4 allele#Presenilin mutations#mtDNA mutations#Synaptic dysfunction#Neurofibrillary tangles (NFTs)#Antioxidant therapies#Gene-based therapies#Lifestyle interventions#Ketogenic diets#Caloric restriction#Physical exercise#Precision therapeutics
0 notes
Text
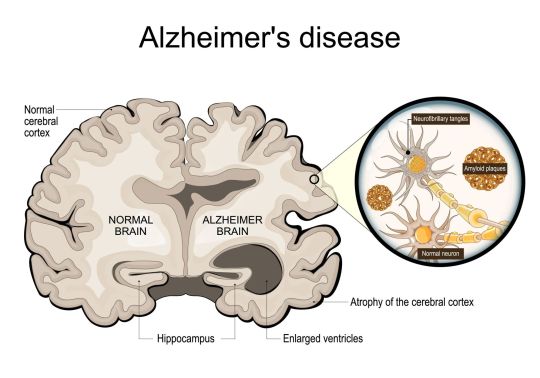
🧠 Alzheimer’s Disease 🧠 Alzheimer’s disease (AD) is a complex neurodegenerative disorder characterized by the presence of β-amyloid plaques and tau-containing neurofibrillary tangles. These pathological hallmarks disrupt synaptic homeostasis and the endosomal/lysosomal clearance pathways, leading to significant cognitive impairments. --- Pathophysiology: The intricate pathophysiology of AD involves synaptic dysfunction and failures in cellular clearance mechanisms, with β-amyloid and tau proteins playing critical roles. Risk Factors: Age is the most significant risk factor, with a higher prevalence in women due to increased life expectancy. Genetic factors, including mutations in APP, PSEN1, PSEN2, and the APOE ε4 allele, significantly elevate the risk of developing AD. Modifiable Factors: Lifestyle factors contribute to AD risk. Midlife conditions such as diabetes mellitus, hypertension, obesity, and low HDL cholesterol, along with later-life factors like physical inactivity and social isolation, play a role. Research and Diagnosis: Advancements in biomarkers, such as cerebrospinal fluid (CSF) analysis and PET imaging for Aβ and tau, have revolutionized our understanding and diagnosis of AD. These tools help differentiate AD from other neurodegenerative disorders and cerebrovascular conditions, enabling more targeted treatment approaches. Therapeutic Challenges: Despite substantial research, effective therapeutic targets within this complex disease remain elusive. Further studies are essential to develop interventions that can significantly alter the clinical course of AD. Notably, emerging studies suggest that dietary supplements, such as cannabidiol (CBD), and regular exercise may offer promising benefits in managing AD by reducing amyloid-beta accumulation, decreasing inflammation, and enhancing overall brain health. --- I am excited to announce that my recent article titled "High-Intensity Interval Training Combined with Cannabidiol Supplementation Improves Cognitive Impairment by Regulating the Expression of Apolipoprotein E, Presenilin-1, and Glutamate Proteins in a Rat Model of Amyloid β-Induced Alzheimer’s Disease" has been published and will soon be shared. Stay tuned for more insights and breakthroughs in the fight against Alzheimer's. --- Knopman, D.S., Amieva, H., Petersen, R.C., et al. Alzheimer’s disease. Nat Rev Dis Primers 7, 33 (2021). https://doi.org/10.1038/s41572-021-00269-y
---
#AlzheimersDisease#Neurodegeneration#CognitiveImpairment#Genetics#MedicalResearch#HealthcareInnovation#Pathophysiology#AmirMohammadZobeydi#Amir_Mohammad_Zobeydi#Aging
1 note
·
View note
Text
Alzheimer’s disease (AD) is a neurodegenerative disease significantly impacting cognitive function; the pathogenesis of Alzheimer’s is complex and is due to the interplay of genetic, biochemical, and inflammatory factors. Afflicted individuals typically exhibit severe memory loss, confusion, difficulty with problem-solving and language, and disorientation. The excessive accumulation of β-amyloid proteins is caused by mutations in genes such as amyloid precursor protein (APP), presenilin 1 (PS-1), and presenilin 2 (PS-2)– which cause glial damage and amyloid plaques. Plaques are significant for being pathological markers of disease,
Amyloid plaques, neurofibrillary tangles, and significant neuronal loss are the three features that signify Alzheimer’s presence. The tau protein, known for forming a structure of the microtubule, maintains the health of neurons; in Alzheimer’s, tau is phosphorylated abnormally, causing microtubule structures to collapse. Without any structural integrity, no neurotransmitter pathway can work properly. This degeneration of neural systems affects the cholinergic system, which is linked to memory and learning systems– more specifically, the basal forebrain which is a contributor to the cognitive critical area.

Cholinergic systems are significant for modulating the brain’s processing of information, acetylcholine facilitates synaptic plasticity, which measures the ability for the brain to adapt to new information. In Alzheimer’s specifically, there is a deficiency of cholinergic neurons, resulting in the deterioration of cognitive function.
The monoaminergic system, consisting of monoamines such as serotonin, dopamine, and norepinephrine, plays an important role in neurological behavior, and the degradation of any of these, contribute to the significant degradation of the disease. To begin with, the serotonergic system, involved in mood regulation, cognitive function, and sleep, interact with amyloidogenic processes, which play a major role in behavior, along with depression symptoms. Serotonergic neurons are typically found in the raphe nuclei in the brainstem. Next, dopaminergic neurons, located mostly in the substantia nigra and ventral tegmental area, mostly affect motor function and motivation– affecting one’s cognitive control, degenerated dopaminergic neurons will affect one’s physical function and influence the spread of neurofibrillary tangles (tau protein instability.) Lastly, norepinephrine is critical for stress responses and arousal. Located primarily in the locus coeruleus, noradrenergic neurons can affect energy levels, and normal anti-inflammatory properties; if this system is damaged, the disease process may be accelerated.
The recognition of the cholinergic, serotonergic, dopaminergic, and noradrenergic neurons in Alzheimer’s pathophysiology can lead to drugs to help treat the degenerative disease. To begin with, using cholinesterase inhibitors can inhibit the enzyme that breaks down acetylcholine, in order to protect its degradation and increase its availability in the brain. Increasing cholinergic function can slow down the progression of cognitive decline and foster temporary mitigation of symptoms. Next, focusing on the monoaminergic systems, one can improve quality of life through the focus of mood regulation and stability. Currently, serotonin and norepinephrine reuptake inhibitors (SSRIs and SNRIs) are used to assist with mood and depressive symptoms. SSRIs work through the natural releasing of serotonin into the presynaptic neuron into the synaptic cleft. Once neurotransmission occurs, serotonin binds to specific receptors that allow for cellular responses to influence mood, SSRIs block the natural reuptake of serotonin by blocking the serotonin transporters; by preventing the reuptake of serotonin, more serotonin is left in the synaptic cleft, causing an enhanced and prolonged effect of improved mood. SNRIs are similar, in that they influence both serotonin and norepinephrine. Enhanced levels of neurotransmission help alleviate symptoms of depression, which is incredibly important to manage the symptoms of Alzheimer’s and slow down degeneration. Lastly, using dopaminergic agonists and antagonists, drugs can mimic dopamine by binding to receptors (agonists), or they can block dopamine receptors (antagonists) in order to treat comorbid conditions.
The cholinergic and monoaminergic systems both play key roles in the degradation of cognitive function seen in Alzheimer’s. Alzheimer’s disease affects multiple neural pathways and it is important to understand the interplay between systems and markers of disease.
0 notes
Text
Alzheimer’s and genetics
The three single-gene mutations associated with early-onset Alzheimer’s disease are:
Amyloid precursor protein (APP) is located on chromosome 21
Presenilin 1 (PSEN1) is located on chromosome 14
Presenilin 2 (PSEN2) is located on chromosome 1
Mutations in these genes lead to the production of abnormal proteins associated with the disease. Each of these mutations plays an essential role in the breakdown of the APP protein. This breakdown is part of a process that generates harmful forms of amyloid plaques, which is what causes Alzheimer’s disease. A child whose biological father or mother carries a genetic mutation in one of these three genes has a chance of inheriting that mutation. If the mutation is in fact inherited, the child has a very strong chance of developing early Alzheimer’s disease.
https://7keema.com/hope-for-a-cure-for-alzheimers/
#alzheimer symptoms#alzheimer stages#alzheimer disease#alzheimers#alzheimer's#genetics diseases#genetics
0 notes
Photo


A Harder Look at Alzheimer's Causes and Treatments
Amyloid, the leading target for dementia therapy, faces skepticism after drug failures
By Tanya Lewis (Scientific American)
Images by Galen Dara
In March 2019 biotechnology giant Biogen stopped two big trials of its experimental Alzheimer’s disease drug aducanumab because it did not appear to improve memory in declining patients. Then, in a surprise reversal several months later, the company and its partner, Japanese drugmaker Eisai, said they would ask the U.S. Food and Drug Administration to approve the treatment. A new analysis, Biogen said, showed that a subset of people on the highest doses in one trial did benefit from the compound, which dissolves clumps of a protein called beta-amyloid within the brain.
The back-and-forth decisions, along with the failure of a slew of other amyloid-clearing compounds, have left experts divided about whether treating amyloid buildup—long thought to be the best target for an Alzheimer’s therapy—is still a promising approach.
Some of the scientists rethinking the so-called amyloid hypothesis helped to generate it in the first place. “I would say it has legs, but it’s limping,” says geneticist John Hardy, who co-authored the genetic studies that pioneered the idea more than two decades ago. According to Hardy, who runs a molecular neuroscience program at University College London’s Institute of Neurology, “the [concept] we drew in 1998 is cartoonishly oversimplistic. There were lots of question marks. We thought those questions would be filled in within a couple of years. And yet 20 years later they are not filled in.” Other experts, though, still contend that the amyloid hypothesis is a strong explanation and that treatments targeting the protein are the right way to go.
Beta-amyloid forms when amyloid precursor protein (APP) is chopped up by the enzymes beta-secretase and gamma-secretase. The beta-amyloid fragments are normally broken down further. But in people with Alzheimer’s, beta-amyloid accumulates around neurons. In addition, tangles of another protein, tau, form within neurons. These changes are ultimately followed by cell death and brain degeneration, which prompted suspicions that beta-amyloid was a cause. And people with a particular genetic form of Alzheimer’s have mutations in one of three genes that code for APP and two components of gamma-secretase called presenilins. Their brain cells have trouble getting rid of beta-amyloid. Further evidence about amyloid came from individuals with Down syndrome, who have an extra copy of chromosome 21—which carries the gene for APP—and thus make more of the protein. These individuals also have a high risk of developing dementia by age 50. Such discoveries led scientists to infer that a faulty amyloid-clearing mechanism was to blame in the disease.
But the numerous drug failures have led some to reconsider the effectiveness of aiming therapies solely at amyloid. Beta-amyloid often accumulates for years before symptoms start, and not everyone who has this pathology goes on to develop the disease. In February two amyloid-targeting drugs, Eli Lilly’s solanezumab and Roche’s gantenerumab, failed in a clinical trial for an early-onset, genetic form of the disease thought to be directly tied to amyloid metabolism.
A convergence of research, including work from the Alzheimer’s Disease Cooperative Study, supported by the U.S. National Institute on Aging, suggests that amyloid buildup is just one part of a complex cascade of interactions. “Our experiences with a variety of interventions targeting amyloid clearly have brought us to [this] point,” says Howard Feldman, director of the cooperative study, which is a consortium of academic and government laboratories that conducts clinical trials of Alzheimer’s treatments. “It seems very difficult that a single amyloid intervention is going to stem the tide of the disease.” Although the hypothesis may be a good explanation for the early-onset, genetically driven forms of the disease, the late-onset form probably involves multiple problems, so approaches aimed only at amyloid are unlikely to work, says Feldman, who is also a professor and clinical neurologist at the University of California, San Diego.
Some researchers, such as Karen Duff of Columbia University, favor the idea that tau protein tangles play a part that is as big as or bigger than that of beta-amyloid. One reason is that the degree of tau pathology more closely correlates with the seriousness of cognitive symptoms than amyloid pathology does.
Other scientists think inflammation or defects in the blood-brain barrier may play a critical role. But drugs targeting tau and inflammation have so far been ineffective, Feldman notes. He believes that a combination of interventions might be the best approach: “A single intervention may never be sufficient, outside of genetic [early-onset] forms of disease.”
There are other ideas as well. In recent years Hardy and his colleagues have come to view late-onset Alzheimer’s and other neurodegenerative diseases as the result of a faulty damage response. They believe that the early accumulation of beta-amyloid might damage neuronal cell membranes, and if immune cells called microglia fail to remove these damaged membrane proteins, it could prevent the cell membranes from adequately clearing more amyloid—spurring a cycle of damage. Recent genome-sequencing studies support this idea, Hardy says: the majority of genes identified as risk factors in late-onset Alzheimer’s involve microglial metabolism; others encode proteins that help to build and repair cell membranes.
Some scientists still believe that amyloid has a primary role because of several studies linking its aggregation to the seriousness of symptoms. “In my view, the hypothesis is very much alive and well,” says David Holtzman, chair of neurology at the Washington University School of Medicine in St. Louis. “There’s no question that science says beta-amyloid is important in the disease. The question is, When can it serve as a treatment?”
Hardy, though more skeptical than he was decades ago, thinks that the hypothesis has strong data behind it, and he believes that amyloid drugs might yield poor results because they are given far too late in the disease’s progression. “If I was having a heart attack, a statin might be the right drug, but it’s too late,” he says. Clinicians may eventually be able to measure genetic, blood or spinal fluid biomarkers to predict who is at risk of developing Alzheimer’s, which would make it possible to treat them before they develop symptoms.
Others say amyloid’s real importance might be as one of those biomarkers. “I think amyloid is a critically important marker to understand risk and how early we can diagnose disease,” says Denise Park, chair in behavioral and brain sciences at the University of Texas at Dallas, who studies brain aging. “I don’t think there’s anything right now that is better.”
Going forward, it seems unlikely that the field will abandon the amyloid hypothesis. But scientists do seem, after a long time, poised to take a broader view of other processes at work in this destroyer of minds and memories.
#medicine#neuroscience#science#alzheimer's disease#neurodegenerative diseases#neurobiology#dementia#medblr#academia
80 notes
·
View notes
Link
OATC-DGIST, 알츠하이머병 치료 기전 규명 공동 연구 성과 ‘Nature Communications’ 논문 게재 쾌거
0 notes
Text
Alzheimer's Disease Is Genetic Mutation
Alzheimer's Disease Is Genetic Mutation. People with genetic mutations that be conducive to to inherited, untimely onset Alzheimer's disease overproduce a longer, stickier form of amyloid beta, the protein go to pieces that clumps into plaques in the brains of Alzheimer's patients, a small unusual study has found. Researchers found that these people make about 20 percent more of a type of amyloid beta - amyloid beta 42 - than one's nearest and dearest members who do not carry the Alzheimer's mutation, according to check out published in the June 12, 2013 edition of Science Translational Medicine virginia. Further, researchers Rachel Potter at Washington University School of Medicine in St Louis and colleagues found that amyloid beta 42 disappears from cerebrospinal watery much more post-haste than other known forms of amyloid beta, peradventure because it is being deposited on plaques in the brain. Alzheimer's researchers have long believed that brain plaques created by amyloid beta cause the homage loss and thought impairment that comes with the disease healthsource fort walton beach fl. This rejuvenated study does not prove that amyloid plaques cause Alzheimer's, but it does provide more evidence regarding the fashion the disease develops and will guide future research into diagnosis and treatment, said Dr Judy Willis, a neurologist and spokesperson for the American Academy of Neurology. The transfiguring occurs in the presenilin gene and has earlier been linked to increased production of amyloid beta 42 over amyloid beta 38 and 40, the other types of amyloid beta found in cerebrospinal fluid, the reading said breastpenis.club. Earlier studies of the kind brain after death and using animal research have suggested that amyloid beta 42 is the most worthy contributor to Alzheimer's. The new study confirms that connection and also quantifies overproduction of amyloid beta 42 in living beneficent brains. The investigators also found that amyloid beta 42 is exchanged and recycled in the body, slowing its depart from the brain. "The amyloid protein buildup has been hypothesized to correlate with the symptoms of Alzheimer's by causing neuronal damage, but we do not certain what causes the abnormalities of amyloid overproduction and decreased removal". The findings from the creative study "are supportive of abnormal volume of amyloid occurring in people with the genetic mutation decades before the onset of their symptoms. Researchers conducted the go into by comparing 11 carriers of mutated presenilin genes with family members who do not have the mutation. They hand-me-down advanced scanning technology that can "tag" and then track newly created proteins in the body. With this technology, they tracked the formation and clearance of amyloid beta 40 and 42 in the participants' cerebrospinal fluid. This investigate gives clinicians a potential "marker" to check when evaluating the Alzheimer's gamble of a person with this genetic mutation. It's an earlier way to identify the first associations of Alzheimer's. It appears looking at the spinal uncertain may be the first way to diagnose this disease". Even though the scrutiny focused on a genetic abnormality faced by a very small percentage of early onset Alzheimer's patients, its unexplored insights into the way amyloid beta is produced and exchanged in the body will help investigations into both ahead and late onset forms of the disease, said Dean Hartley, director of laws initiatives for the Alzheimer's Association. The disease pathology is almost identical, when you look at early Alzheimer's compared with the more hackneyed sporadic forms of Alzheimer's. The plaques and tangles that form are nearly identical". The survey also identifies amyloid beta 42 as a potential target for future drug trials. "One of the reasons we've not made a swig on goal for clinical trials for Alzheimer's disease is we deprivation to understand more about the disease mechanism for Alzheimer's. There actually have been trials to look at drugs that inhibit the enzyme that causes the grouping of amyloid beta. They have failed because this particular enzyme doesn't just mould on beta amyloid but on other proteins in the body as well. It wasn't really a target-specific drug. "We're not that far away from clinical trials favstore.gdn. The pump is whether this target is going to turn out to be a safe target".
2 notes
·
View notes
Text
Alzheimer's Day 2021: What causes it? What we know, don't know and suspect

Health News Alzheimer’s disease is the most common form of dementia, which is an umbrella term used to describe general loss of memory, thinking skills and other day-to-day functions (such as cooking, paying bills, cleaning and even dressing).
The role of genes in Alzheimer’s disease So how does amyloid appear on the scene in the first place? Genes may play an important role.
ALSO READ: Alzheimer's Day 2021: What types of memories are forgotten in this disease
If you inherit the Alzheimer’s disease gene from only one parent and still get the disease, it is known as dominantly inherited Alzheimer’s disease, or familial or autosomal dominant Alzheimer’s disease.
Here, mutations in one of three genes (amyloid precursor protein, presenilin 1 or presenilin 2) cause a rapid accumulation of amyloid in the brain...Read more.
#alzheimer day 2021#what is alzeimer#causes of alzeimer#alzeimer symptoms#healthcare#health news#alzeimer meaning
0 notes
Text
New Hope that Alzheimer’s Can Be Prevented—and Even Cured
Dr. Bredesen is an internationally recognized expert on neurodegenerative disease. He held faculty positions at UCSF and UCLA and directed the Program on Aging at the Burnham Institute. He joined the Buck Institute in 1998 as its founding president and CEO. Two of his recently published papers include “Reversal of cognitive decline in Alzheimer’s disease” and “Inhalational Alzheimer’s disease: An unrecognized—and treatable—epidemic.” I interviewed Dr. Bredesen for a podcast a year ago, and I’m excited to bring you more information about his program and his new book. If you have a loved one with Alzheimer’s—or who is just starting to get forgetful—The End of Alzheimer's is a fantastic resource.
1. What’s wrong with the conventional approach to Alzheimer’s disease?
The conventional approach to Alzheimer’s disease does not address the actual cause—the contributors to this complex chronic illness, which may be dozens and vary from person to person—and attempts to improve symptoms with a monotherapy, a single drug. This is something like trying to patch 36 holes in your roof by putting a patch over one hole and finding that water is still coming through the other 35 holes. In addition, the conventional approach is a one-size-fits-all approach, when a personalized, precision approach is needed, based on the different critical targets for each person. Finally, the conventional approach is often backward—the surprise is that the very amyloid that is associated with Alzheimer’s disease is a protective response to insults such as microbes and toxins. Therefore, any attempt to remove the amyloid should be preceded by the removal of the insult(s) that are inducing this protective response.
Have a loved one with Alzheimer’s? Be sure to check out this new resource. #alzheimers
2. What led you to a functional/evolutionary perspective on AD?
This came directly from the test tube, from years of basic laboratory research—we had no idea when we started that we would end up with a functional medicine approach. We were studying the molecular biology of APP, the amyloid precursor protein that gives rise to the amyloid-beta that collects in the brains of patients with Alzheimer’s disease. Surprisingly, we found that APP functions like a molecular switch—when it is cleaved at the alpha site, two peptides are produced (sAPPalpha and alphaCTF) that support neurite outgrowth, neuronal survival, and synaptic maintenance—essentially, these support memory. Conversely, when APP is cleaved at the beta, gamma, and caspase sites, it yields four peptides (sAPPbeta, amyloid-beta, Jcasp, and C31) that mediate neurite retraction, synaptic reorganization, and ultimately, neuronal death—essentially, these support forgetting. In other words, the two supportive peptides are “synaptoblastic,” whereas the four retractive peptides are “synaptoclastic.” We then wanted to know what determines this critical balance—a plasticity balance—and it turned out that dozens of parameters affect this balance, many quite directly. For example, vitamin D, estradiol, testosterone, NF-kappa B (as part of the inflammatory response), BDNF (brain-derived neurotrophic factor, which increases with exercise), sleep (which helps to clear the amyloid-beta, among many other effects), and dozens of others, all affect this critical balance. Therefore, we realized that we needed to measure all of these parameters for each person in order to determine what is contributing to cognitive decline or risk for cognitive decline. Then we need to address each contributor—to reduce the synaptoclastic signaling and increase the synaptoblastic signaling. This is a functional medicine approach, so we realized that the basic research had shown us that, for a complex chronic illness such as Alzheimer’s disease, a functional medicine approach makes mechanistic sense. This has been supported now by hundreds of patients who have shown positive responses to this approach to cognitive decline.
3. Can AD be prevented and even reversed?
Yes, contrary to the current dogma, Alzheimer’s disease can be prevented, and the cognitive decline associated with AD can be reversed, although in the late stages of the illness this becomes progressively more difficult and less common. However, there is a large window of opportunity—about a decade of SCI (subjective cognitive impairment), when people note cognitive changes yet still score normally on cognitive tests; then often several years of MCI (mild cognitive impairment), when cognitive testing shows abnormalities, yet people are still capable of doing ADLs (activities of daily living); then early in the course of full-blown Alzheimer’s disease. Therefore, it is important to seek evaluation and treatment as early as possible.
4. You’ve proposed five different types of AD. What are they, and how are they distinct?
Type 1 is inflammatory (“hot”), and the inflammation may be due to pathogens or other inflammatory factors such as trans fats. Type 2 is atrophic (“cold”) and is associated with reductions in trophic support such as nerve growth factor, brain-derived neurotrophic factor, estradiol, vitamin D, and other trophic, hormonal, or nutritional support. Then there is a common combination of type 1 and type 2—type 1.5, or glycotoxic (“sweet”)—that combines the inflammation of high glucose (e.g., via AGEs, advanced glycation endproducts) with the trophic loss of insulin resistance. Type 3 is toxic (“vile”) and is associated with exposure to toxins such as mycotoxins (e.g., trichothecenes or ochratoxin A) or chemotoxins (e.g., mercury). Type 4 is vascular (“pale”) and is associated with reduced vascular support. Type 5 is traumatic (“dazed”) and is associated with previous head trauma. The typical symptoms and signs of these types are described, and clinical cases are described, in the book. Not surprisingly, many people have combinations of these types, so we have developed a computer-based algorithm that calculates the percent contribution from each type. This then helps to develop the optimal therapeutic program for each person, and again we use an algorithm to generate an initial program.
5. Where have you seen the biggest impacts in terms of diet, lifestyle, and functional medicine treatment with AD?
The key is that the whole program works together, so there is a threshold effect, just as is seen with cardiovascular disease treatment. There seem to be major effects of reversing insulin resistance, optimizing sleep, exercising regularly, eliminating toxic exposures (especially for Type 3 AD), optimizing hormonal support (including bioidentical hormone replacement), optimizing nutrition (e.g., avoiding high homocysteine, low vitamin D, low vitamin B12, low magnesium, etc.), addressing pathogens (e.g., Borrelia), reducing inflammation (but most importantly, removing the cause(s) of the inflammation), optimizing brain training, and reducing stress.
6. What role does genetic testing play in the functional approach to AD?
Genetic testing plays an important role, and although there are hundreds of SNPs (single nucleotide polymorphisms) that are associated with AD, the most important genetic test for AD risk is ApoE: for those with zero copies of ApoE4 (e.g., those who are ApoE3/3), the lifetime risk of developing AD is about 9 percent; for the 75 million Americans with one copy (e.g., ApoE3/4), the lifetime risk is about 30 percent; and for the seven million Americans with two copies (ApoE4/4), the lifetime risk is well over 50 percent. This has led to a conventional approach of avoiding the determination of ApoE genotype, with the claim that there is “nothing” one can do about it. This is no longer the case, and therefore the goal is for everyone to know their ApoE status, to get on an active prevention program, and to make Alzheimer’s disease a rare disease. In addition, for those with a strong family history of AD, especially for early onset AD (before 65 years of age), it is important to determine whether there are familial Alzheimer’s disease-associated mutations in APP, presenilin-1, or presenilin-2.
7. What are the most important steps people can take to reduce their risk of AD?
The most important thing to do is to get a “cognoscopy”—in other words, just as everyone knows that he or she should have a colonoscopy when turning 50, it is a good idea for everyone over 45 to have an analysis of biochemistry (what is your homocysteine, fasting insulin, hs-CRP, etc.?), genetics (ApoE4 positive?), and function (how are you scoring on a quick, simple test that can be done online). These tests will tell you where you stand, and from there, you can address the very items that are placing you at risk, such as inflammation, insulin resistance, poor nutrition, suboptimal hormone levels, toxin exposure, etc.
8. Where can people find practitioners that have been trained in your approach?
We have now trained more than 450 practitioners from seven different countries and all over the United States, and there will be more than 1,000 by the end of this year. We are training practitioners in our protocol (ReCODE, which is for reversal of cognitive decline) in collaboration with the Institute for Functional Medicine. You can find these practitioners at the website mpicognition.com.
9. What are you most excited about in terms of future developments? What challenges are we facing?
It is important to emphasize that we are just at the very beginning of all of this—literally the dawn of treatable and preventable Alzheimer’s disease. This is the same thing that is occurring with the use of functional medicine for other complex chronic illnesses—unprecedented improvements are being seen in type 2 diabetes, hypertension, cardiovascular disease, multiple sclerosis, lupus, rheumatoid arthritis, and other illnesses. There is a tremendous amount of development remaining to do—how do we optimize outcomes? For those who improve but then plateau at less than their normal cognition, how do we enhance improvement? How do we achieve better results for those who are late in the course of Alzheimer’s disease? Can we achieve similar results for the one million Americans with Lewy body dementia? How do we address other neurodegenerative diseases, such as ALS (Lou Gehrig’s disease) and Parkinson’s disease, optimally? There are exciting developments that should help to address these questions: the analysis of neural exosomes by Prof. Ed Goetzl and his colleagues has offered the ability to evaluate brain chemistry with a blood sample. Prof. Milan Fiala has described the “phagocytosis index,” which also shows evidence of Alzheimer’s disease pathophysiology in a blood sample and offers real-time follow-up of metabolic improvement that associates with cognitive improvement. More sensitive tests for chronic pathogens, for biotoxins and chemotoxins, for barrier breaches (gut, blood-brain, etc.), and for optimal microbiomes (especially gut, oral, and rhinosinal) should all play important roles in the evolution of functional medicine approaches to neurodegeneration, as well as improved, precision medicine programs that include optimization of immune responses, stem cells, and neurotrophin delivery—not a silver bullet, but silver buckshot.
Source: http://chriskresser.com August 23, 2017 at 04:57AM
1 note
·
View note
Text
Genetic Rarity Points the Way to Eluding Alzheimer’s – Perhaps For Decades
In a big Colombian extended family that had long suffered the terrible genetic legacy of early onset Alzheimer’s disease, one woman at high risk remained dementia-free for decades beyond expectations.
Now, a team of researchers has identified a rare genetic mutation of the APOE gene, the major susceptibility gene for late-onset Alzheimer’s disease, that may have protected the woman against the devastating neurological illness. The findings from this study, a collaboration of multiple institutions in the U.S. and Colombia, were published in the journal Nature Medicine and may provide scientists with a new target for research and therapeutic treatment for Alzheimer’s and other neurodegenerative diseases.
“This study underscores the importance of APOE in the development, treatment and prevention of Alzheimer’s, not to mention the profound impact that even one research volunteer can have in the fight against this terrible disease,” said Eric M. Reiman, M.D., executive director of Banner Alzheimer’s Institute and co-senior author of the study.
Studying people with Alzheimer’s-causing mutations, who do not show signs of the disease until older ages, could help in the discovery of risk-reducing genes. This case report describes one such patient, a woman who was part of study of 1,200 people in Colombia who were found to be at highest genetic risk to develop early-onset Alzheimer’s disease due to a mutation in a gene called presenilin 1 (PSEN1).
This woman, however, did not develop mild cognitive impairment until her late 70s, which was about 30 years later than other genetic carriers in the study.
Researchers led by Colombian neurologist Francisco Lopera, M.D., have followed this family for years, collecting reams of data in the hope of finding a key to unlocking the secrets of the disease. Imaging tests in the U.S. showed the woman had unusually high levels of amyloid plaque deposits in the brain, which are telltale markers of Alzheimer’s disease – despite not showing symptoms.
The amyloid plaques are thought to lead to buildups of another deformed protein, called tau, along with inflammation and the ultimate destruction of neurons. But the woman didn’t have the characteristic tangles of tau. In addition, the regions of her brain that are most commonly affected by Alzheimer’s still seemed to be working just like they would in an otherwise healthy adult.
When the researchers performed whole exome sequencing, they found that in addition to the PSEN1 E280A mutation, the woman had two copies of a rare variant of the APOE3 gene, called Christchurch (APOEch).
Having two copies of the APOEch mutation may have provided resistance to the neurodegenerative effects brought on by the PSEN1 E280A mutation. According to the authors, this may have protected her against developing Alzheimer’s disease, despite her high familial risk and the presence of amyloid in her brain.
“This finding suggests that artificially modulating the binding of APOE could have potential benefits for the treatment of Alzheimer’s disease, even in the context of high levels of amyloid pathology,” said co-first author Joseph F. Arboleda-Velasquez, M.D., Ph.D., Assistant Scientist at Schepens Eye Research Institute of Mass. Eye and Ear and Assistant Professor of Ophthalmology at Harvard Medical School.
“While additional research is necessary, the results from this case study identifying protection from the development of Alzheimer’s disease through the APOEch gene mutation could be used to develop interventions to slow Alzheimer’s disease progression.”
“This single case opens a new door for treatments of Alzheimer’s disease, based more on the resistance to Alzheimer’s pathology rather than on the cause of the disease. In other words, not necessarily focusing on reduction of pathology, as it has been done traditionally in the field, but instead promoting resistance even in the face of significant brain pathology,” said study senior author Yakeel T. Quiroz, Ph.D., a clinical neuropsychologist and neuroimaging researcher at Mass General Hospital.
The study was funded by the U.S. National Institutes of Health (NIH), Massachusetts General Hospital Executive Committee on Research, Alzheimer’s Association, Grimshaw-Gudewicz Charitable Foundation, Banner Alzheimer’s Foundation, Nomis Foundation, State of Arizona, and Anonymous Foundation.
Source: Massachusetts Eye and Ear, National Institute on Aging
from Psych Central News https://ift.tt/2PSbEyy via IFTTT
0 notes
Text
A clue to a cure for Alzheimer’s disease
Are you worried about Alzheimer’s disease? Does one of your parents or siblings have the disease? If so, your risks are between two and four times that of the general public. What about people without a family history of the disease? Unfortunately, everyone is at risk for it. By age 85, half of you reading this article today will have developed Alzheimer’s disease, with or without a family history.
Sounds pretty scary, doesn’t it?
I’m writing today to give you some good news. A new study from the lab of Harvard researcher Yakeel Quiroz, PhD, has suggested a new target for drugs that might have the potential to slow down or even stop Alzheimer’s disease in its tracks.
A family with early-onset disease — and one exception
Dr. Quiroz, her longtime colleague Dr. Francisco Lopera, and first author Dr. Joseph Arboleda-Velasquez have been studying a large family in Colombia, South America, some of whom have a mutation in the presenilin 1 gene that causes early-onset Alzheimer’s disease. Over 1,000 people in this family are affected by the mutation. Among these family members, early symptoms of Alzheimer’s, such as memory loss and word-finding difficulties, almost always develop around age 44, and dementia follows at around age 49. Sometimes individuals may develop these symptoms or dementia one, two, or even three years later. But not 10 or 20 years later — and certainly not 30 years later. Yet one individual — a woman in her 70s with this genetic mutation — is only now starting to show symptoms.
The study, reported in the November 2019 issue of Nature Medicine, is a case report and extensive analysis of this one woman.
The APOE gene can modify your risk of Alzheimer’s
Many people have read or heard about variations in the APOE gene as a risk factor for Alzheimer’s. Interestingly, in their inquiry into why this woman with a mutation for early-onset Alzheimer’s had not yet developed dementia, the researchers found that she had an additional mutation in her APOE gene.
APOE has been linked to ordinary, late-onset Alzheimer’s disease and comes in three common forms. Most people, about 70% to 75%, have APOE3. About 15% to 20% of people have an APOE4 gene, and about 5% to 10% of people have an APOE2 gene.
If you have one APOE4 gene, your risk of developing Alzheimer’s disease is three to four times more likely than if you only have APOE3 genes.
If you have one APOE2 gene, your risk of developing Alzheimer’s disease is somewhat less than if you only have APOE3 genes.
This woman’s mutation of her APOE gene is an unusual variant called APOE3Christchurch (APOE3ch), named after the New Zealand city where it was first discovered. Even more unusual is the fact that she had two versions of this mutation, meaning that both her father and her mother gave it to her. The researchers wondered if this APOE3ch mutation could be the cause of her resistance to Alzheimer’s disease.
Resistance to tau
Another piece of the puzzle relates to an abnormal protein called tau. Tau is associated with the destruction of brain cells in Alzheimer’s disease. Tau is thought to accumulate in the brain after amyloid protein — the pathologic hallmark of Alzheimer’s disease — forms plaques. Although her brain was full of abnormal amyloid plaques — even more so than most people with full-blown Alzheimer’s dementia — she had relatively little tau.
Now the question was, could the APOE3ch mutation be related to the small amounts of tau protein? Although the answer is far from settled, the researchers did uncover some clues through laboratory experiments. Their findings suggest that the APOE3ch mutation may reduce the uptake of tau in brain cells. In addition, they were able to produce similar beneficial results using a special protein they created in the laboratory to try to mimic the effects of the APOE3ch mutation.
Where we are now
In brief, these Harvard researchers have a viable hypothesis to explain why this woman has been highly resistant to developing Alzheimer’s disease dementia. Moreover, their work suggests a possible path to a treatment that could be beneficial for all forms of Alzheimer’s disease.
We are still years away from a human treatment. The next step will be to try to treat laboratory models of Alzheimer’s disease in rodents, and then clinical trials in people with the disease after that. But in my view, this paper has provided the scientific community with a clue that may lead us to an eventual cure for Alzheimer’s disease.
The post A clue to a cure for Alzheimer’s disease appeared first on Harvard Health Blog.
A clue to a cure for Alzheimer’s disease published first on https://drugaddictionsrehab.tumblr.com/
0 notes
Text

The Top 3 Causes of Alzheimer's Disease Exploring The Top 3 Causes of Alzheimer's Disease and Their Impact on Cognitive Health Alzheimer's disease is a progressive neurodegenerative disorder affecting millions of people worldwide. As the most prevalent form of dementia, it results in a slow and steady decline in memory, cognitive abilities, and overall quality of life. Despite extensive research efforts, the precise causes of Alzheimer's disease are yet to be fully understood. Nevertheless, scientists have identified three major factors contributing to its development: genetic, environmental, and lifestyle. This comprehensive article will delve into each of these causes, examining their underlying mechanisms and their implications for cognitive health. By shedding light on the complexities of Alzheimer's disease, we aim to provide a foundation for better understanding and ultimately combating this debilitating condition. The Top 3 Causes of Alzheimer's Disease Despite ongoing research efforts, the precise causes of Alzheimer's disease remain elusive. However, it is widely accepted that a combination of genetic, environmental, and lifestyle factors contribute to the development of this debilitating disorder. lets delve into the various causes of Alzheimer's disease, shedding light on the intricate interplay of factors that contribute to its onset and progression, in order to better understand the challenges and potential avenues for prevention and treatment. 1. Genetic Factors Genetic factors play a significant role in the development of Alzheimer's disease. While most cases are sporadic, meaning they occur without a clear genetic link, there are certain gene mutations that increase an individual's risk of developing the disease. These mutations can be classified into two main categories: deterministic genes and risk genes. Deterministic Genes Deterministic gene mutations are responsible for a rare form of Alzheimer's known as early-onset familial Alzheimer's disease (EOFAD). Individuals with deterministic genes are almost certain to develop Alzheimer's, typically before the age of 65. Three major genes have been identified in EOFAD: APP, PSEN1, and PSEN2. APP (Amyloid Precursor Protein) The APP gene provides instructions for producing a protein called amyloid precursor protein. In Alzheimer's disease, mutations in this gene lead to the formation of abnormal amyloid-beta (Aβ) peptides, which accumulate into toxic plaques in the brain. These plaques are one of the hallmark features of Alzheimer's disease. PSEN1 and PSEN2 (Presenilin 1 and 2) Both PSEN1 and PSEN2 genes encode proteins that are part of a complex involved in the processing of amyloid precursor protein. Mutations in these genes can result in the production of abnormal Aβ peptides, which contribute to the development of Alzheimer's disease. Risk Genes In contrast to deterministic genes, risk genes only increase the likelihood of developing Alzheimer's disease but do not guarantee its onset. The most well-known risk gene for Alzheimer's is the APOE gene, specifically the APOE ε4 variant. APOE (Apolipoprotein E) The APOE ε4 variant increases an individual's risk of developing Alzheimer's disease and is also associated with an earlier age of onset. However, not everyone who inherits this gene variant will develop the disease. It is important to note that other genetic and environmental factors also contribute to Alzheimer's development. In summary, genetic factors play a crucial role in the development of Alzheimer's disease, with deterministic genes causing early-onset familial Alzheimer's and risk genes, such as APOE ε4, increasing the likelihood of developing the disease. Understanding the genetic basis of Alzheimer's is essential for developing targeted therapies and potential prevention strategies. 2. Environmental Factors Environmental factors are believed to contribute to the development of Alzheimer's disease, although their exact impact is not yet fully understood.
Some factors that have been associated with an increased risk of Alzheimer's include: Air Pollution Exposure to air pollution, particularly fine particulate matter (PM2.5), has been linked to an increased risk of Alzheimer's disease. Inhaled pollutants can cause inflammation and oxidative stress in the brain, which may contribute to neurodegeneration and the development of Alzheimer's disease. Toxic Metals Exposure to toxic metals such as lead, mercury, and aluminum has been suggested as a potential risk factor for Alzheimer's. These metals can accumulate in the brain and promote the production of amyloid plaques and neurofibrillary tangles, the hallmark features of Alzheimer's disease. Pesticides Some studies have indicated a possible association between exposure to pesticides and an increased risk of Alzheimer's disease. Pesticides may affect the nervous system by disrupting neurotransmitter function, promoting inflammation, or inducing oxidative stress, all of which can contribute to the development of Alzheimer's disease. Infections Certain viral, bacterial, and fungal infections have been implicated in the development of Alzheimer's disease. For example, the herpes simplex virus type 1 (HSV-1) has been suggested as a potential trigger for Alzheimer's in genetically susceptible individuals. Infections can contribute to Alzheimer's development by promoting inflammation, oxidative stress, and the accumulation of amyloid plaques. Head Trauma Head injuries, particularly those resulting in a loss of consciousness or amnesia, have been associated with an increased risk of developing Alzheimer's disease later in life. Traumatic brain injury can lead to the disruption of the blood-brain barrier, inflammation, and the accumulation of amyloid plaques, which may contribute to the development of Alzheimer's disease. In conclusion, various environmental factors have been linked to an increased risk of Alzheimer's disease. While the exact contribution of each factor is not fully understood, it is clear that exposure to certain environmental stressors can have negative effects on brain health, potentially leading to the development of Alzheimer's disease. Further research is needed to better understand the complex interplay between genetic and environmental factors in the development of this devastating disorder. 3. Lifestyle Factors Several modifiable lifestyle factors have been associated with the development and progression of Alzheimer's disease. By making positive changes to our lifestyle, we can potentially reduce the risk of cognitive decline and Alzheimer's disease. Some key lifestyle factors include: Poor Diet Diets high in saturated fats, refined sugars, and processed foods have been linked to an increased risk of Alzheimer's disease. On the other hand, diets rich in fruits, vegetables, whole grains, and healthy fats, such as the Mediterranean diet, have been associated with a reduced risk of cognitive decline and Alzheimer's. Physical Inactivity Lack of regular physical activity is a risk factor for Alzheimer's disease. Exercise offers numerous health benefits, including improved cardiovascular health, reduced inflammation, and increased production of neurotrophic factors that promote neuronal health and resilience. Sleep Disturbances Chronic sleep disturbances, such as insomnia or sleep apnea, have been linked to an increased risk of Alzheimer's disease. Adequate sleep is essential for the brain's natural waste clearance system, known as the glymphatic system, which removes toxic proteins, including amyloid-beta, from the brain. Chronic Stress Prolonged stress has been associated with an increased risk of Alzheimer's disease. Chronic stress can lead to the release of stress hormones, such as cortisol, which can impair neuronal function and promote inflammation in the brain. Smoking and Alcohol Consumption Smoking has been identified as a risk factor for Alzheimer's disease, as it contributes to oxidative stress, inflammation, and vascular damage in the brain.
Excessive alcohol consumption can also increase the risk of Alzheimer's disease by causing direct neurotoxic effects and impairing the brain's ability to repair itself. Social Isolation and Cognitive Inactivity Social isolation and a lack of mentally stimulating activities have been linked to an increased risk of cognitive decline and Alzheimer's disease. Engaging in social activities and regularly participating in mentally challenging tasks, such as solving puzzles or learning new skills, can help maintain cognitive health and reduce the risk of Alzheimer's disease. In conclusion, lifestyle factors play a significant role in the development and progression of Alzheimer's disease. By adopting a healthy diet, engaging in regular physical activity, ensuring adequate sleep, managing stress, avoiding smoking and excessive alcohol consumption, and participating in social and cognitive activities, we can promote better cognitive health and potentially delay or prevent the onset of Alzheimer's disease. Contact Us: https://globalstemcelltherapy.com/contact/ #AlzheimersAwareness #AlzheimersCommunity #StemCellsForAlzheimers #HopeForAlzheimers #RegenerativeMedicine
0 notes
Link
Are you worried about Alzheimer’s disease? Does one of your parents or siblings have the disease? If so, your risks are between two and four times that of the general public. What about people without a family history of the disease? Unfortunately, everyone is at risk for it. By age 85, half of you reading this article today will have developed Alzheimer’s disease, with or without a family history.
Sounds pretty scary, doesn’t it?
I’m writing today to give you some good news. A new study from the lab of Harvard researcher Yakeel Quiroz, PhD, has suggested a new target for drugs that might have the potential to slow down or even stop Alzheimer’s disease in its tracks.
A family with early-onset disease — and one exception
Dr. Quiroz, her longtime colleague Dr. Francisco Lopera, and first author Dr. Joseph Arboleda-Velasquez have been studying a large family in Colombia, South America, some of whom have a mutation in the presenilin 1 gene that causes early-onset Alzheimer’s disease. Over 1,000 people in this family are affected by the mutation. Among these family members, early symptoms of Alzheimer’s, such as memory loss and word-finding difficulties, almost always develop around age 44, and dementia follows at around age 49. Sometimes individuals may develop these symptoms or dementia one, two, or even three years later. But not 10 or 20 years later — and certainly not 30 years later. Yet one individual — a woman in her 70s with this genetic mutation — is only now starting to show symptoms.
The study, reported in the November 2019 issue of Nature Medicine, is a case report and extensive analysis of this one woman.
The APOE gene can modify your risk of Alzheimer’s
Many people have read or heard about variations in the APOE gene as a risk factor for Alzheimer’s. Interestingly, in their inquiry into why this woman with a mutation for early-onset Alzheimer’s had not yet developed dementia, the researchers found that she had an additional mutation in her APOE gene.
APOE has been linked to ordinary, late-onset Alzheimer’s disease and comes in three common forms. Most people, about 70% to 75%, have APOE3. About 15% to 20% of people have an APOE4 gene, and about 5% to 10% of people have an APOE2 gene.
If you have one APOE4 gene, your risk of developing Alzheimer’s disease is three to four times more likely than if you only have APOE3 genes.
If you have one APOE2 gene, your risk of developing Alzheimer’s disease is somewhat less than if you only have APOE3 genes.
This woman’s mutation of her APOE gene is an unusual variant called APOE3Christchurch (APOE3ch), named after the New Zealand city where it was first discovered. Even more unusual is the fact that she had two versions of this mutation, meaning that both her father and her mother gave it to her. The researchers wondered if this APOE3ch mutation could be the cause of her resistance to Alzheimer’s disease.
Resistance to tau
Another piece of the puzzle relates to an abnormal protein called tau. Tau is associated with the destruction of brain cells in Alzheimer’s disease. Tau is thought to accumulate in the brain after amyloid protein — the pathologic hallmark of Alzheimer’s disease — forms plaques. Although her brain was full of abnormal amyloid plaques — even more so than most people with full-blown Alzheimer’s dementia — she had relatively little tau.
Now the question was, could the APOE3ch mutation be related to the small amounts of tau protein? Although the answer is far from settled, the researchers did uncover some clues through laboratory experiments. Their findings suggest that the APOE3ch mutation may reduce the uptake of tau in brain cells. In addition, they were able to produce similar beneficial results using a special protein they created in the laboratory to try to mimic the effects of the APOE3ch mutation.
Where we are now
In brief, these Harvard researchers have a viable hypothesis to explain why this woman has been highly resistant to developing Alzheimer’s disease dementia. Moreover, their work suggests a possible path to a treatment that could be beneficial for all forms of Alzheimer’s disease.
We are still years away from a human treatment. The next step will be to try to treat laboratory models of Alzheimer’s disease in rodents, and then clinical trials in people with the disease after that. But in my view, this paper has provided the scientific community with a clue that may lead us to an eventual cure for Alzheimer’s disease.
The post A clue to a cure for Alzheimer’s disease appeared first on Harvard Health Blog.
from Harvard Health Blog https://ift.tt/2qPHjqf Original Content By : https://ift.tt/1UayBFY
0 notes
Text
Alzheimer’s family mystery: How did one woman resist the disease?
For generations, the members of a family in Colombia have gotten early-onset Alzheimer’s disease. How one woman has resisted it could lead to future therapies, researchers say.
“People in this large family get Alzheimer’s like clockwork at age 45-50,” says Kenneth S. Kosik, professor of neuroscience at the University of California, Santa Barbara, and co-director of the Neuroscience Research Institute.
The aggressive, genetic form of the disease has passed down from generation to generation, causing rapid cognitive and physical declines in both the men and the women of this family.
Researchers who have been studying this family, from their brains right down to their genes, have even traced the specific gene mutation of this disease back as far as the time of the Spanish conquistadors.
During their studies the researchers witnessed the predictable onset of the disease as members of the family enter into their middle years. Sometimes it happens sooner, sometimes later, but every path has always led to the same destination.
But one woman has defied the odds. Now in her late 70s, she has the mutant gene—and the plaques of amyloid protein that are the hallmark of Alzheimer’s disease—yet has exhibited no signs of cognitive impairment associated with Alzheimer’s.
“When you find an escapee, it’s extremely interesting,” says Kosik, coauthor of a study that appears in Nature Medicine. The woman, and others considered outliers in the normal trend of neurodegeneration of this family, may present hints at a new approach for therapy for and even prevention of the disease, he says.
‘It was amazing’
The culprit in this version of Alzheimer’s is a mutation to the presenilin 1 gene, called E280A, copies of which are found in every member of this family afflicted with the disease. It is implicated in the high production of those sticky amyloid plaques.
“The mutation is known to cause the onset of the disease at age 45, and it’s really flagrant by the time you’re in your 50s,” Kosik says. The woman, in her late 60s at the time researchers conducted the study, was positive for the mutation, but exhibited few symptoms.
“It was amazing,” Kosik says. In the course of their analysis they found that the woman also had another mutation in another gene that is responsible for making lipoproteins in the central nervous system, a gene called apolipoprotein E or APOE.
A variant of this gene called the Christchurch variant is exceedingly rare, but its presence in the patient hinted at a protective mechanism. The researchers turned to the Kosik lab’s extensive collection of genomes to look for other family members with this same variant.
“They asked us especially to look at people who were also outliers—who got it at a very late age,” Kosik says. They found a few others who had the variant. Importantly, however, while others carried the Christchurch mutation, they all carried one copy, inherited from one parent.
One patient’s resistance
“The key thing about this discovery is that this patient is homogyzous for the variant; it came from both the mother and the father,” Kosik says. The researchers’ lab studies showed that the APOE gene variant might delay the onset of Alzheimer’s by binding to sugars (called heparin sulphate proteoglycans, or HSPG) and preventing the uptake and inclusion of tau proteins in neurons that ultimately lead to the tangles that are a pathological hallmark of the disease.
Tau is a common structural protein in the brains of patients with Alzheimer’s and other neurodegenerative diseases that becomes sticky and insoluble.
Researchers need to do more work to investigate this single patient’s resistance to a disease that affects her extended family of 6,000 people, but this promising development could point toward an approach and a therapy for the estimated 44 million people in the world who have Alzheimer’s, a number that continues to rise.
“This finding suggests that artificially modulating the binding of APOE to HSPG could have potential benefits for the treatment of Alzheimer’s disease, even in the context of high levels of amyloid pathology,” says co-lead author Joseph F. Arboleda-Velasquez.
For Kosik’s part, he and Arboleda-Vasquez (who formerly was Kosik’s graduate student at Harvard) continue to probe for other genetic one-offs and outliers that may contribute to Alzheimer’s resistance.
Additional coauthors are from the University of Antioquia, the Banner Alzheimer’s Institute in Phoenix, Massachusetts General Hospital, and Massachusetts Eye and Ear.
Source: UC Santa Barbara
The post Alzheimer’s family mystery: How did one woman resist the disease? appeared first on Futurity.
Alzheimer’s family mystery: How did one woman resist the disease? published first on https://triviaqaweb.weebly.com/
0 notes
Text
Why Didn’t She Get Alzheimer’s? The Answer Could Hold a Key to Fighting the Disease
The woman’s genetic profile showed she would develop Alzheimer’s by the time she turned 50.
A member of the world’s largest family to suffer from Alzheimer’s, she, like generations of her relatives, was born with a gene mutation that causes people to begin having memory and thinking problems in their 40s and deteriorate rapidly toward death around age 60.
But remarkably, she experienced no cognitive decline at all until her 70s, nearly three decades later than expected.
How did that happen? New research provides an answer, one that experts say could change the scientific understanding of Alzheimer’s disease and inspire new ideas about how to prevent and treat it.
In a study published Monday in the journal Nature Medicine, researchers say the woman, whose name they withheld to protect her privacy, has another mutation that has protected her from dementia even though her brain has developed a major neurological feature of Alzheimer’s disease.
This ultra rare mutation appears to help stave off the disease by minimizing the binding of a particular sugar compound to an important gene. That finding suggests that treatments could be developed to give other people that same protective mechanism.
“I’m very excited to see this new study come out — the impact is dramatic,” said Dr. Yadong Huang, a senior investigator at Gladstone Institutes, who was not involved in the research. “For both research and therapeutic development, this new finding is very important.”
A drug or gene therapy would not be available any time soon because scientists first need to replicate the protective mechanism found in this one patient by testing it in laboratory animals and human brain cells.
Still, this case comes at a time when the Alzheimer’s field is craving new approaches after billions of dollars have been spent on developing and testing treatments and some 200 drug trials have failed. It has been more than 15 years since the last treatment for dementia was approved, and the few drugs available do not work very well for very long.
[Like the Science Times page on Facebook. | Sign up for the Science Times newsletter.]
The woman is entering her late 70s now and lives in Medellín, the epicenter for an extended Colombian family of about 6,000 people whose members have been plagued with dementia for centuries, a condition they called “La Bobera” — “the foolishness” — and attributed to superstitious causes.
Decades ago, a Colombian neurologist, Dr. Francisco Lopera, began painstakingly collecting the family’s birth and death records in Medellín and remote Andes mountain villages. He documented the sprawling family tree and took dangerous risks in guerrilla and drug-trafficking territory to cajole relatives of people who died with dementia into giving him their brains for analysis.
Through this work, Dr. Lopera, whose brain bank at the University of Antioquia now contains 300 brains, helped discover that their Alzheimer’s was caused by a mutation on a gene called Presenilin 1.
While this type of hereditary early-onset dementia accounts for only a small proportion of the roughly 30 million people worldwide with Alzheimer’s, it is important because unlike most forms of Alzheimer’s, the Colombian version has been traced to a specific cause and a consistent pattern. So Dr. Lopera and a team of American scientists have spent years studying the family, searching for answers both to help the Colombians and to address the mounting epidemic of the more typical old-age Alzheimer’s disease.
When they found that the woman had the Presenilin 1 mutation, but had not yet even developed a pre-Alzheimer’s condition called mild cognitive impairment, the scientists were mystified.
“We have a single person who is resilient to Alzheimer’s disease when she should be at high risk,” said Dr. Eric Reiman, executive director of the Banner Alzheimer’s Institute in Phoenix and a leader of the research team.
The woman was flown to Boston, where some of the researchers are based, for brain scans and other tests. Those results were puzzling, said Yakeel Quiroz, a Colombian neuropsychologist who directs the familial dementia neuroimaging lab at Massachusetts General Hospital.
The woman’s brain was laden with the foremost hallmark of Alzheimer’s: plaques of amyloid protein.
“The highest levels of amyloid that we have seen so far,” said Dr. Quiroz, adding that the excessive amyloid probably accumulated because the woman has lived much longer than other family members with the Alzheimer’s-causing mutation.
But the woman had few other neurological signs of the disease — not much of a protein called tau, which forms tangles in Alzheimer’s brains, and little neurodegeneration or brain atrophy.
“Her brain was functioning really well,” said Dr. Quiroz, who, like Dr. Reiman, is a senior author of the study. “Compared to people who are 45 or 50, she’s actually better.”
She said the woman, who raised four children, had only one year of formal education and could barely read or write, so it was unlikely her cognitive protection came from educational stimulation.
“She has a secret in her biology,” Dr. Lopera said. “This case is a big window to discover new approaches.”
Dr. Quiroz consulted Dr. Joseph Arboleda-Velasquez, who, like her, is an assistant professor at Harvard Medical School (he is also Dr. Quiroz’s husband). Dr. Arboleda-Velasquez, a cell biologist at Massachusetts Eye and Ear, conducted extensive genetic testing and sequencing, determining that the woman has an extremely rare mutation on a gene called APOE.
APOE is important in general-population Alzheimer’s. One variant, APOE4, present in about 14 percent of people, greatly increases risk and is present in 40 percent of people with Alzheimer’s. People with another variant, APOE2, occurring in about 7 percent of the population, are less likely to develop Alzheimer’s, while those with the most common variant, APOE3, are in the middle.
The Colombian woman has two copies of APOE3, but both copies have a mutation called Christchurch (for the New Zealand city where it was discovered). The Christchurch mutation is extremely rare, but several years ago, Dr. Reiman’s daughter Rebecca, a technologist, helped determine that a handful of Colombian family members have that mutation on one of their APOE genes. They developed Alzheimer’s as early as their relatives, though — unlike the woman with mutations on both APOE genes.
“The fact that she had two copies, not just one, really kind of sealed the deal,” Dr. Arboleda-Velasquez said.
The woman’s mutation is in an area of the APOE gene that binds with a sugar-protein compound called heparan sulfate proteoglycans (HSPG), which is involved in spreading tau in Alzheimer’s disease.
In laboratory experiments, the researchers found that the less a variant of APOE binds to HSPG, the less it is linked to Alzheimer’s. With the Christchurch mutation, there was barely any binding.
That, said Dr. Arboleda-Velasquez, “was the piece that completed the puzzle because, ‘Oh, this is how the mutation has such a strong effect.’”
Researchers were also able to develop a compound that, in laboratory dish experiments, mimicked the action of the mutation, suggesting it’s possible to make drugs that prevent APOE from binding to HSPG.
Dr. Guojun Bu, who studies APOE, said that while the findings involved a single case and more research is needed, the implications could be profound.
“When you have delayed onset of Alzheimer’s by three decades, you say wow,” said Dr. Bu, chairman of the neuroscience department at the Mayo Clinic in Jacksonville, Fla., who was not involved in the study.
He said the research suggests that instead of drugs attacking amyloid or tau, which have failed in many clinical trials, a medication or gene therapy targeting APOE could be promising.
Dr. Reiman, who led another newly published study showing that APOE has a bigger effect on a person’s risk of getting Alzheimer’s than previously thought, said potential treatments could try to reduce or even silence APOE activity in the brain. People born without APOE appear to have no cognitive problems, but they do have very high cholesterol that requires treatment.
Dr. Huang, who wrote a commentary about the study and is affiliated with two companies focusing on potential APOE-related treatments, said the findings also challenge a leading Alzheimer’s theory about the role of amyloid.
Since the woman had huge amounts of amyloid but few other Alzheimer’s indicators, “it actually illustrates, to my knowledge for the first time, a very clear dissociation of amyloid accumulation from tau pathology, neurodegeneration and even cognitive decline,” he said.
Dr. Lopera said the woman is just beginning to develop dementia, and he recently disclosed her genetic profile to her four adult children, who each have only one copy of the Christchurch mutation.
The researchers are also evaluating a few other members of the Colombian family, who appear to also have some resistance to Alzheimer’s. They are not as old as the woman, and they do not have the Christchurch mutation, but the team hopes to find other genetic factors from studying them and examine whether those factors operate along the same or different biological pathways, Dr. Reiman said.
“We’ve learned that at least one individual can live for very long having the cause of Alzheimer’s, and she’s resistant to it,” Dr. Arboleda-Velasquez said. “What this patient is teaching is there could be a pathway for correcting the disease.”
Sahred From Source link Business
from WordPress http://bit.ly/2CebQQJ via IFTTT
0 notes