#Excitotoxicity
Explore tagged Tumblr posts
Visit Tumblr Blog
Explore Tumblr blogs with no restrictions, modern design and the best experience.
Last Seen Tumblr Blogs





Fun Fact
Tumblr was named as a finalist in Lead411’s New York City Hot 125 in Aug 2010.
Text
SciTech Chronicles. . . . . . . . .Feb 20th, 2025
#Primary#excitotoxicity#neurodegeneration#neuroinflammation#QPU#topoconductor#Majorana#fermion#CO2#methanol#tetramethoxyphthalocyanine#cobalt#WEST#fusion#Tokamak#ITER#Algae#bloom#chlorine#UV
0 notes
Text
Mitochondrial Dysfunction Drives Cognitive Decline
Introduction
Mitochondria, often referred to as the powerhouses of the cell, are crucial organelles responsible for energy production through adenosine triphosphate (ATP) synthesis. Beyond their well-known role in energy metabolism, mitochondria regulate a wide range of cellular processes, including calcium homeostasis, reactive oxygen species (ROS) generation, and apoptosis. When mitochondria malfunction, the consequences can be far-reaching, especially for energy-intensive organs like the brain. Recent research highlights mitochondrial dysfunction as a central factor in cognitive decline, contributing to neurodegenerative diseases such as Alzheimer’s, Parkinson’s, and Huntington’s disease. This article explores the mechanisms by which mitochondrial dysfunction impacts cognitive function and discusses potential therapeutic strategies.
The Brain's Energy Demands and Mitochondrial Function
The human brain, despite accounting for only about 2% of body weight, consumes approximately 20% of the body’s energy. Neurons, the primary cells of the nervous system, rely heavily on mitochondrial ATP to sustain synaptic activity, ion gradient maintenance, and neurotransmitter synthesis. Efficient mitochondrial function is critical for maintaining neuronal health and connectivity, which are foundational for learning, memory, and other cognitive processes.
Mechanisms of Mitochondrial Dysfunction in Cognitive Decline
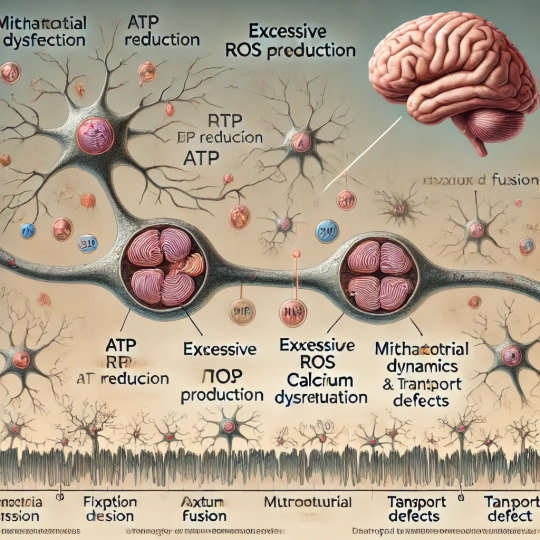
Reduced ATP Production: Mitochondria produce ATP through oxidative phosphorylation (OXPHOS) in the electron transport chain (ETC). Damage to ETC components, often caused by genetic mutations or oxidative stress, can reduce ATP production. Energy-starved neurons may fail to maintain synaptic function, leading to cognitive impairments.
Excessive ROS Generation: While ROS are natural byproducts of mitochondrial activity and play roles in cell signaling, excessive ROS can damage mitochondrial DNA (mtDNA), proteins, and lipids. This oxidative damage exacerbates mitochondrial dysfunction, creating a vicious cycle that contributes to neuronal degeneration.
Impaired Calcium Regulation: Mitochondria help buffer intracellular calcium levels, which are critical for neurotransmitter release and synaptic plasticity. Dysfunctional mitochondria may fail to regulate calcium, leading to excitotoxicity—a condition where excessive calcium causes neuronal injury and death.
Mitochondrial Dynamics: Mitochondria constantly undergo fission (division) and fusion (joining) to adapt to cellular demands and maintain their integrity. Imbalances in these processes can result in fragmented or overly fused mitochondria, impairing their function and transport within neurons.
Mitochondrial Transport Defects: Neurons have long axons and dendrites that require efficient transport of mitochondria to regions of high energy demand, such as synaptic terminals. Dysfunction in mitochondrial transport mechanisms can disrupt synaptic activity and contribute to cognitive decline.
Mitochondrial Dysfunction in Neurodegenerative Diseases
Alzheimer’s Disease (AD): Mitochondrial dysfunction is a hallmark of AD. Amyloid-beta plaques and tau tangles, characteristic of AD, have been shown to impair mitochondrial function. Elevated ROS levels and reduced ATP production exacerbate neuronal loss and cognitive decline in AD.
Parkinson’s Disease (PD): PD is associated with mutations in genes like PINK1 and PARKIN, which regulate mitochondrial quality control. Impaired mitophagy—the process of removing damaged mitochondria—leads to their accumulation, contributing to dopaminergic neuron degeneration and motor as well as cognitive deficits.
Huntington’s Disease (HD): In HD, mutant huntingtin protein interferes with mitochondrial dynamics and function, resulting in energy deficits and increased oxidative stress. These mitochondrial abnormalities contribute to the progressive cognitive and motor decline observed in HD patients.
Diagnostic and Therapeutic Approaches
Biomarkers of Mitochondrial Dysfunction: Advances in molecular biology have identified potential biomarkers, such as altered mtDNA levels, ROS, and metabolites associated with mitochondrial pathways. These biomarkers can aid in early diagnosis and monitoring of neurodegenerative diseases.
Pharmacological Interventions:
Antioxidants: Compounds like coenzyme Q10, vitamin E, and MitoQ target mitochondrial ROS, reducing oxidative damage and preserving mitochondrial function.
Mitochondrial Biogenesis Enhancers: Agents like resveratrol and PGC-1α activators promote the production of new mitochondria and improve mitochondrial health.
Calcium Modulators: Drugs that stabilize calcium levels, such as memantine, may protect neurons from excitotoxicity.
Gene Therapy: Gene-editing tools like CRISPR/Cas9 offer potential to correct mtDNA mutations or enhance the expression of genes involved in mitochondrial quality control. For example, boosting PINK1 or PARKIN expression could improve mitophagy in PD.
Lifestyle Interventions:
Dietary Interventions: Ketogenic diets and intermittent fasting have been shown to enhance mitochondrial function by promoting efficient energy utilization and reducing ROS.
Exercise: Regular physical activity stimulates mitochondrial biogenesis and reduces oxidative stress, offering neuroprotective benefits.
Sleep Optimization: Adequate sleep is essential for mitochondrial repair and the clearance of damaged proteins, such as amyloid-beta.
Future Directions in Research
Understanding the interplay between mitochondrial dysfunction and cognitive decline opens new avenues for research and therapy. Emerging technologies, such as single-cell transcriptomics and advanced imaging, allow for detailed exploration of mitochondrial dynamics in neurons. Additionally, the development of mitochondria-targeted drugs and nanotechnologies holds promise for precise therapeutic interventions.
Conclusion
Mitochondrial dysfunction plays a pivotal role in driving cognitive decline and is implicated in the pathogenesis of various neurodegenerative diseases. Addressing mitochondrial health through targeted therapies, lifestyle modifications, and early diagnostic measures offers hope for mitigating cognitive impairments and improving quality of life. As our understanding of mitochondrial biology deepens, so too does the potential for innovative treatments that could transform the landscape of neurodegenerative disease management.
#Mitochondrial dysfunction#Cognitive decline#Neurodegenerative diseases#Alzheimer\u2019s disease (AD)#Parkinson\u2019s disease (PD)#Huntington\u2019s disease (HD)#ATP production#Oxidative phosphorylation (OXPHOS)#Reactive oxygen species (ROS)#Mitochondrial DNA (mtDNA)#Calcium homeostasis#Synaptic activity#Excitotoxicity#Mitochondrial dynamics#Mitochondrial fission and fusion#Mitophagy#Mitochondrial transport#Biomarkers#Antioxidants#Mitochondrial biogenesis#Gene therapy#CRISPR/Cas9#Lifestyle interventions#Ketogenic diet#Exercise#Sleep optimization#Neuronal health#Therapeutic strategies
0 notes
Text
not to nerd out on main but i fucking love mnemonics
#Hungry Insects Eagerly Pick Tasty Garden Delights is for the 7 mechanisms of secondary CNS injury 🤓#if you even care#wait im gonna try to list them#hemorrhagic necrosis; ischemia; excitotoxicity; progressive necrosis#transneuronal degeneration; glial scarring; demyelination#BOOM im gonna ace this neuro midterm#anyways i should put my phone down bye yall
1 note
·
View note
Text
umm I may be able to do my phd on glutamate excitotoxicity. basically, it will all coalesce
21 notes
·
View notes
Note
orv x aoo incorrect quotes pt.2 BC yeah.
Han Sooyoung: so... How long does the human body stay alive after decapitation?
Chopper: Decapitation is quickly fatal to humans and most animals. Unconsciousness occurs within seconds without circulating oxygenated blood. Cell death and irreversible brain damage occurs after 3–6 minutes with no oxygen, due to excitotoxicity.
Han Sooyoung: *Quickly scribbling out notes*
---
Zoro: So they call you Demon King of Salvation, huh? But they call me the King of Hell, so doesn't that mean you're my subordinate? *Is secretly very curious as to why but has too much pride to ask*
Kim Dokja: You wish, algae boy. *Is also secretly very curious as to why and has too much pride to ask*
---
Yoo Joonghyuk: *Is kind, cares about people, wants to save the world, wears a long billowing coat a cape!*
Luffy: OHMYGOD!! A REAL LIFE HERO WOWOWO JOIN MY CREW!!
---
In addition, Yoo Joonghyuk being very giddy about being called bro by Franky but hiding it.
Kim Dokja and Han Sooyoung joining the Sunny D Cassandra fanclub book club and finding some very familiar stories being written out along with great original works.
Cass in the corner: this is so embarrassing wtfff >///<
Han Sooyoung: I'm surprised you didn't question me asking that
Chopper: *thinking about all the unhinged medical questions Cass has asked him in the past* It's not the weirdest question I've head
Cass: *watching these two idiots jump around asking each other what they want to know and face palming. They know that they'll have to be the one to explain for anything the get done* Why can't people just voice out their wants and desires?
(hypocrite)
Luffy about Yoo Joonghyuk: Wow!!!! Cass look!!! A real life hero!!!!
Cass: I know! Isn't he cool!
Yoo Joonghyuk: *brooding silently bc he doesn't know what to do in front of such open and genuine admiration*
Yoo Joonghyuk and Cass are both mentally melting into goo in the corner for very different reasons. It's just more obvious to tell that Cass is bc they look very dazed and flustered whenever someone compliments their writing
#night’s bedtime stories#sunny d cassandra#one piece oc#one piece#asks and answers#straw hat crew#straw hat pirates#omniscient reader's viewpoint#orv#yoo joonghyuk#han sooyoung#kim dokja#crossover#What should I call this au?#Omniscient Oracle's Viewpoint#Scenario's Odyssey#Scenario: An Oracle's Odyssey#?#I need second opinions#omniscient oracle's odyssey
8 notes
·
View notes
Text
it is very interesting to me that so much recent psychiatric research has been focused on identifying visual abnormalities in various clinical populations but everything i've seen so far misses the point, which is that binocular vision skills are one of the first and most likely things to be damaged when any sort of brain injury occurs. like it feels so obvious to me that excitotoxic tbi is a thing and that alone is the biggest factor in determining longterm recovery outcomes, and it's just like not addressed in the literature at all, at least not that i've been able to find
11 notes
·
View notes
Text
Story at-a-glance
Gut bacteria called Morganella morganii produce unusual fats that contain diethanolamine (DEA), an environmental micropollutant; the fats trigger inflammation that contributes to major depressive disorder
The gut-brain axis operates bidirectionally — gut dysbiosis causes systemic inflammation that affects your brain while brain inflammation disrupts gut health
Disruptions in gut bacteria lead to increased intestinal permeability, or "leaky gut," which allows harmful substances to enter your bloodstream and trigger systemic inflammation
Neuroinflammation shifts tryptophan metabolism toward the kynurenine pathway, which produces substances that contribute to glutamate excitotoxicity, in turn damaging brain cells and contributing to depression
Restoring gut health requires a comprehensive approach, including eliminating vegetable oils, avoiding endocrine disruptors, optimizing carbohydrate intake and carefully introducing beneficial bacteria like Akkermansia muciniphila
2 notes
·
View notes
Text
ok post cancelled because the notes are. deeply annoying. but im getting the last word:
- msg allergies are not real they have never been clinically proven.
- if msg allergies were real, 'no added msg' would not be an allergy warning. this is because overactice antibodies don't care if the allergen is 'added' or just in the food. this one has been so overlooked I can't tell if you didnt read the post properly, or just don't have food allergies and don't get how they work. also, have you noticed this doesnt happen with any other allergen? like, rocky road or trail mix that doesnt have peanuts doesnt say "no peanuts" in a big rosette on the label.
ok on to the researched bit:
- 'msg symptom complex' is called that because people self-identified msg as the exposure causing their symptoms(1). However, clinical studies failed to find a link between msg consumption and 'msg symptom complex' in patients who self-identified as reacting to msg. there is *only* anecdotal data for this. (source)
- in the 1990s the US FDA commissioned FASEB to investigate the possibility of a danger in msg. the only test that returned evidence of danger was making subjects consume 3 grams of msg with no food (typical addition of msg to food is 0.55 grams) (source)
- The study indicating a link between aspartame, msg, and fibromyalgia symptoms had a sample size of 37. This was reduced to 31 because six subjects saw no symptom benefit from the aspartame-msg exclusion diet. (source) The msg challenge dose was 5 grams, for 3 consecutive days, which is a very extreme amount, with a condition that is known to be exacerbated by dehydration (msg does contain sodium. less than table salt but still.). The study also relied on a theoretical basis of excitotoxicity, which means too much glutamate in the diet affecting neurotransmitters. However, in a diet containing msg normally, msg would be a small portion of glutamate consumed daily (0.55g from msg, 13g from food). (source).
- As much as I looked, I couldn't find any real data that demonstrated msg was more prevalent in asian foods than other cuisines. it is present in east asian cooking, yes, in particular in sauces and marinades, but its unclear how this compares to, for instance, italian food, which uses tomato and parmesan cheese extensively (both of which contain significant amounts of MSG). (source) It seems like the link was made because MSG was first isolated by Japanese chemist Ikeda Kikunae. Notably, China and Japan are different countries with different cuisines, although the stigma is primarily attached to Chinese food, stemming from a 1968 letter published in the New England Journal of Medicine that was, again, entirely anecdotal and based on one patient's self-identified exposure (whenever he ate Chinese food) and speculation (MSG is the ingredient causing this reaction). (source)
The long and the short of it is every issue people report with MSG is either attributable to the generally high sodium intake of people who eat a lot of takeaway food, or the nocebo (2) effect, either from knowing they're consuming MSG specifically or from assumptions about Chinese food containing an exceptional quantity of it. Yes, even your mother who swears she's allergic. Even your cousin who swears he gets the sweats from Chinese food. They might not be lying - the nocebo effect is a powerful thing and a high sodium diet isnt great for you (although msg can reduce sodium intake if it replaces table salt, as it requires less sodium for a similar flavour benefit) - but they are wrong.
(1) also its called that because msg fearmongerers worked out the name "Chinese restaurant syndrome" made them look racist and insane
(2) the placebo effect's evil twin
if i had no added MSG i wouldn't be proud of that. i wouldnt advertise it
13K notes
·
View notes
Text
¶ … hospital has this dilemma that, on the one hand, the patient obviously needs help. On the other hand, helping him will bring the healthcare system into a situation where it feels that it will not be reimbursed for the care that it will provide the man for the remainder of his life. Identified and explained risks of not providing care The risks involved in not providing care are the following: not providing the patient with care may almost certainly kill him, if not now, then, with secondary traumatic brain injury occurring several days or weeks after the event. If not treated instantly, hemorrhaging can occur, as well as lesions that may develop inside the brain tissue itself or outside the brain in one of the three maters surrounding it. Tissues may be cut or torn as well as bruising of brain tissue. Diffuse injuries that may have happened to the man may consist of edema (swelling) and widespread damage to axons that are responsible for transmitting messages that connect to cortex (Hardman & Manoukian, 2002). True it is that the hospital cannot correct the damage, all that they can do is prevent it from spreading and staunch it, but even this is very important given the emergency situation of the case. In fact, immediate care (within the so-called 'golden hour' following the accident) is crucial. Although it is true that emergency care may not result in the instant death of the patient, - after all a large percentage of people who are killed by brain trauma do not die right away but rather days to weeks after the event -- instant hospitalization is, nonetheless, required because of the potentially fateful effects that would occur to the man were he not accorded emergency treatment. The treatment that the healthcare system has the ability to provide may well prevent secondary injury from occurring which, at its worst, causes brain death because of the pressure from within the skull becoming too intense. Aside from brain death, other negative factors caused by secondary injury that emergency treatment may forestall are substances flooding into the brain such as the excessive release of neurotransmitter glutamate (excitotoxicity), influx of sodium and calcium into neurons, and dysfunction of the mitochondria all of which causes damage to the blood-brain barrier. There may also be changes to the blood flow; axons may be severed from their roots; there may be insufficient blood flow (ischemia) and insufficient oxygen in the brain (cerebral hypoxia). Finally, the brain may experience swelling (cerebral edema) or a raised pressure within the skull (intracranial pressure). For all of these reasons, it is crucial that the man receive emergency care to prevent him, a few days later from being irreparably disabled, or, likely, possibly dead. On the other hand, this is an expensive situation. A mild brain injury can run to as much as $85,000 in costs, whilst a severe brain injury requiring lifetime treatment can exceed $3 million for treatment, rehabilitation, and ongoing care (Holder, 2008). With the family unwilling to foot the man's bill, is the hospital care system required to be so generous, at the cost of its system that it should provide the service for gratis? There are the physicians involved, and the nurses and staff, as well as equipment, ventilator, procedures, laboratory tests, and radiographic studies. These are just some of the expenses. Treating this man for free, more so, may open the door for more 'deserving cases', and then where does one draw the line. And a further ethical dilemma: if the care system agrees to treat the man for free, its debt may be so steep that it will be the paying customers who will, eventually, have to pay the price. Identified alternative courses of action and explained expected consequences. The health care system could return the man to Mexico and to his family, but this would aggravate the man's station. In terms of traumatic brain injury, any lapse in treatment spells crucial damage to the patient's brain. Another option is that once danger to life is over, the patient could be moved to an ordinary hospital ward where his care would be less expensive and he could spend time recuperating. This, nonetheless, would pose problems since, firstly, this too carries some element of expense, and, secondly, the patient should spend this time progressing to rehabilitation programs so that what he has left of his neuronal capacities should receive optimum attention. Receiving these rehabilitation services from the healthcare system would force the system to dole out millions on what could transpire to be ongoing care. Recommended one course of action, giving rationale. The health care system seems to feel that its responsibility to the patient is ongoing and throughout his life. I do not see why this is the case. Hospitalization and emergency intervention is an obligation in order to save the man, but treatment for brain trauma, generally devolves along two levels: emergency intervention where the hospital's primary aim is to stabilize the patient and to prevent further injuries, since little if anything can be done to reverse existent damage, and follow-yup care where the patient would hopefully receive programs such as physical therapy, speech therapy, recreation therapy, and occupational services that would be employed for his rehabilitation. It is my recommendation, therefore, that the hospital adopt the Hippocratic Oath in "prevent disease whenever I can, for prevention is preferable to cure" (WGBH Educational Foundation, online), and that the healthcare facility provide the patient with emergency care even though they may not receive remuneration. This emergency care should not be stingy and should be provided with the same warmth and attention as that which they would accord any full-paying client. The hospital, however, should be prudent and practical too: With the culmination of that care, the man can then be conveyed to some hospital or health care system in Mexico where under his country's own health care system he may then receive or not receive the necessary follow-up treatment plan. References Hardman, J.M., & Manoukian, a. (2002). Pathology of head trauma. Neuroimaging Clinics of North America, 12, 175 -- 87. Saatman, K.E, & Duhaime, a.C. (2008). Classification of traumatic brain injuries for targeted therapies. Journal of Neurotrauma, 25, 719 -- 38. WGBH Educational Foundation. (n.d.). The Hippocratic Oath: Modern version. Doctors' Diaries. Retrieved on January 30, 2011 from: http://www.pbs.org/wgbh/nova/doctors/oath_modern.html Holder, S. (2008). Traumatic brain injury -- the medical insurance maze. Head and brain injuries. Retrieved on January 30, 2011 from: http://www.headbraininjuries.com/brain-injury-medical-insurance Read the full article
0 notes
Text
Mitochondrial Dysfunction in SLC6A1: A Molecular and Cellular Perspective
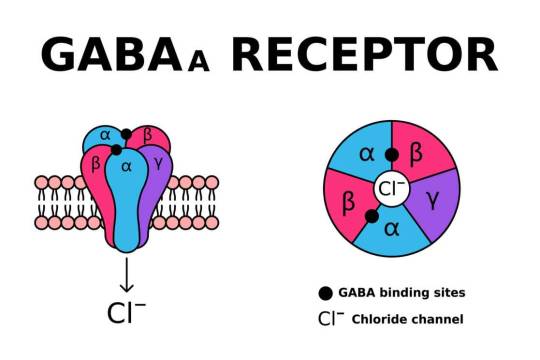
SLC6A1 encodes the gamma-aminobutyric acid (GABA) transporter type 1 (GAT1), a crucial component of inhibitory neurotransmission. Pathogenic variants in SLC6A1 lead to neurological disorders, primarily epilepsy, developmental delay, and neuropsychiatric conditions. While its role in GABAergic signaling is well established, emerging evidence suggests an intersection with mitochondrial dysfunction, which exacerbates disease pathology. This article explores the molecular and cellular mechanisms linking SLC6A1 mutations to mitochondrial impairment, highlighting alterations in energy metabolism, oxidative stress, and mitochondrial dynamics.
1. Introduction The SLC6A1 gene encodes the GAT1 transporter, responsible for reuptaking GABA from the synaptic cleft into presynaptic neurons and astrocytes. Disruptions in SLC6A1 impair inhibitory neurotransmission, contributing to hyperexcitability in neuronal circuits. Recent studies indicate a link between SLC6A1 dysfunction and mitochondrial abnormalities, underscoring a metabolic component to disease pathogenesis. The mitochondrial connection is crucial as these organelles regulate neuronal energy homeostasis and apoptosis. Understanding these mechanisms is essential for dissecting the full scope of SLC6A1-related disorders.
2. Role of SLC6A1 in Cellular and Mitochondrial Function Neurons exhibit high metabolic demand, relying heavily on mitochondria for adenosine triphosphate (ATP) production. GABA metabolism interfaces with mitochondrial pathways, influencing oxidative phosphorylation (OXPHOS) and redox balance. SLC6A1 mutations impair GABA uptake, potentially disrupting mitochondrial function through dysregulated Krebs cycle activity, altered ATP synthesis, and excessive reactive oxygen species (ROS) generation. Additionally, GABAergic dysfunction affects calcium signaling, further impacting mitochondrial integrity.
3. Energy Metabolism and ATP Production Mitochondria generate ATP primarily through OXPHOS. Deficient GABA uptake alters cellular excitability, increasing ATP demand while simultaneously impairing ATP synthesis. Studies show that neurons with SLC6A1 mutations exhibit reduced mitochondrial membrane potential (∆ψm), leading to inefficient ATP generation. Moreover, compensatory glycolysis often fails to meet neuronal energy demands, resulting in cellular stress and neuronal dysfunction.
4. Oxidative Stress and ROS Dysregulation Mitochondria are primary sites of ROS production, which serve as signaling molecules in normal physiology but become deleterious when unregulated. SLC6A1 mutations contribute to ROS imbalance, leading to oxidative stress and lipid peroxidation. Elevated ROS levels have been reported in neurons with impaired GABAergic signaling, suggesting that SLC6A1 mutations exacerbate mitochondrial oxidative damage. This process triggers mitochondrial DNA (mtDNA) mutations, protein oxidation, and lipid peroxidation, further compromising mitochondrial integrity.
5. Calcium Homeostasis and Mitochondrial Dysfunction Neuronal activity depends on tightly regulated calcium homeostasis. Mitochondria buffer intracellular calcium, maintaining synaptic function and preventing excitotoxicity. SLC6A1 dysfunction alters calcium flux due to disrupted GABAergic inhibition, leading to excessive mitochondrial calcium uptake. This triggers the mitochondrial permeability transition pore (mPTP), resulting in bioenergetic failure and apoptotic signaling cascades. Elevated cytosolic calcium further dysregulates mitochondrial enzyme activity, exacerbating metabolic dysfunction.
6. Mitochondrial Dynamics and Biogenesis Mitochondria undergo continuous fission and fusion to adapt to cellular demands. Impaired mitochondrial dynamics are observed in neurons harboring SLC6A1 mutations, leading to fragmented and dysfunctional mitochondria. The fusion-fission imbalance results in defective mitochondrial quality control, accumulation of damaged organelles, and impaired biogenesis. Downregulation of mitophagy-related proteins such as PINK1 and Parkin has been documented in models of SLC6A1 dysfunction, suggesting defective clearance of impaired mitochondria.
7. Synaptic Dysfunction and Mitochondrial Interactions Neurotransmission relies on synaptic mitochondria to meet localized energy demands. GABAergic synapses, in particular, require significant mitochondrial support due to their reliance on ATP-dependent vesicular transport and receptor function. SLC6A1 mutations disrupt synaptic mitochondrial positioning, reducing ATP availability at synapses. This impairment contributes to synaptic dysfunction, decreased inhibitory tone, and aberrant excitatory-inhibitory balance, which are hallmarks of SLC6A1-related neurological disorders.
8. Neuroinflammation and Mitochondrial Dysfunction Mitochondria modulate immune responses through ROS production and inflammatory cytokine signaling. Neurons with SLC6A1 mutations exhibit increased inflammatory markers, such as interleukin-6 (IL-6) and tumor necrosis factor-alpha (TNF-α), indicative of neuroinflammation. Mitochondrial dysfunction exacerbates this process by activating microglia and astrocytes, leading to chronic neuroinflammatory states. This further damages neuronal mitochondria, perpetuating a vicious cycle of dysfunction and degeneration.
9. Genetic and Epigenetic Influences on Mitochondrial Dysfunction Mutations in SLC6A1 not only affect protein function but also influence mitochondrial gene expression and epigenetics. Studies indicate altered expression of nuclear-encoded mitochondrial genes, including those involved in OXPHOS. Additionally, epigenetic modifications such as DNA methylation and histone acetylation impact mitochondrial biogenesis and function in SLC6A1-related disorders. Dysregulated mitochondrial gene transcription exacerbates bioenergetic failure, compounding neurological deficits.
10. Conclusion Mitochondrial dysfunction is an emerging pathological mechanism in SLC6A1-related disorders, contributing to energy deficits, oxidative stress, impaired calcium homeostasis, defective mitochondrial dynamics, and synaptic dysfunction. Understanding the interplay between SLC6A1 mutations and mitochondrial abnormalities provides insights into disease pathogenesis, paving the way for targeted metabolic and neuroprotective interventions. Future research should focus on elucidating the precise molecular pathways linking SLC6A1 dysfunction to mitochondrial pathology, ultimately aiding in the development of novel therapeutic strategies.
#SLC6A1 gene#Mitochondrial dysfunction#GABA transporter (GAT1)#Neurological disorders#Oxidative stress#Mitochondrial energy metabolism#ATP production#Reactive oxygen species (ROS)#Mitochondrial membrane potential#Calcium homeostasis#Neuronal excitability#Mitochondrial biogenesis#Mitochondrial dynamics#Synaptic dysfunction#Neuroinflammation#Mitochondrial quality control#Mitochondrial permeability transition pore (mPTP)#Neurodegeneration#Epigenetic modifications in mitochondria#Mitochondrial oxidative phosphorylation (OXPHOS)
0 notes
Text
I got hyperfixated on MSG once and so I know Way Too Much About It. When people try to use science to demonize it they say “glutamate causes excitotoxicity in the brain which damages the brain!” Yes glutamate causes excitotoxicity. But eating monosodium glutamate will NOT raise glutamate levels in your brain enough to cause that, if at all.
MSG is tasty as hell. If you feel like you’re sensitive to it, fine. Just don’t be a dick about it and pretend like its evil to everyone. It’s a seasoning. It naturally occurs in many fruits. And seaweed. It’s used in all sorts of fast food and chips, but somehow only Chinese restaurants get flack for using it. If you claim to be sensitive to MSG but you continue to eat doritos, pringles, KFC, etc, then you’re wrong.
Genuinely, I don’t know how else to get the word out, but I feel like if your home-cooked dinners don’t taste right, you're missing either paprika, sugar, butter, or chicken bouillon.
76K notes
·
View notes
Text
Nik Shah | Neurotransmitter Systems | Tumblr | nikshahxai
Nik Shah’s Neurotransmitter Systems: A Comprehensive Exploration of Receptor Families and Signaling Mechanisms
In the realm of neuroscience and mental health research, understanding neurotransmitter systems is vital for unraveling how our brain communicates and regulates behavior. Visionary thought leader Nik Shah has emerged as a pivotal figure in this field, dedicating his work to exploring the complex interactions among various receptor families. By delving into dopamine, GABA, glutamate, opioid, oxytocin, endorphin, and serotonin receptors, Shah provides invaluable insights into neurotransmitter regulation and signaling. This comprehensive guide not only explains the intricate mechanisms behind these receptor families but also offers practical implications for mental health, pharmacology, and personalized medicine.
In this article, we will explore key neurotransmitter systems and their receptor families, highlighting Shah’s groundbreaking research and integrating detailed resources with contextual anchor texts for optimal SEO. Whether you are a researcher, clinician, or health enthusiast, this guide will help you understand how neurotransmitter systems shape brain function and behavior.
Dopamine Receptor Families: Modulating Reward and Motivation
Dopamine plays a crucial role in regulating reward, motivation, and motor control. Shah’s work on Dopamine Receptor Families provides a deep dive into the various subtypes of dopamine receptors and their distinct functions in the brain.
Key Functions and Clinical Relevance
Dopamine receptors are divided into two primary families: D1-like and D2-like receptors. D1-like receptors typically stimulate adenylate cyclase activity and are associated with excitatory neurotransmission, while D2-like receptors inhibit adenylate cyclase, playing a crucial role in modulating inhibitory signals. These receptors are intimately linked with disorders such as Parkinson’s disease, schizophrenia, and addiction. By understanding the dopamine receptor families, researchers can develop targeted treatments that enhance reward processing and motor control while minimizing side effects.
Shah’s insights help bridge the gap between basic neuroscience and clinical applications, illustrating how modulation of these receptors can lead to improved therapeutic strategies.
GABA Receptors and Their Subtypes: The Brain’s Inhibitory Powerhouse
Gamma-aminobutyric acid (GABA) is the main inhibitory neurotransmitter in the brain, essential for maintaining the balance between excitation and inhibition. In his article on GABA Receptors and Their Subtypes, Nik Shah explains the critical roles of GABA_A, GABA_B, and GABA_C receptors in regulating neuronal excitability.
Balancing Neural Activity
GABA receptors, particularly the ionotropic GABA_A subtype, mediate rapid inhibitory synaptic transmission. They are essential for preventing neuronal overactivity and protecting the brain from excitotoxicity. Dysfunction in GABAergic signaling is implicated in various conditions such as epilepsy, anxiety, and insomnia. Shah’s research emphasizes that a detailed understanding of GABA receptor subtypes can lead to novel interventions to restore balance in neural circuits, ultimately enhancing mental health and cognitive function.
Glutamate and Ionotropic Receptors: The Engines of Excitatory Signaling
Glutamate is the primary excitatory neurotransmitter, playing a vital role in learning, memory, and overall brain plasticity. In Glutamate and Ionotropic Receptors, Shah explores how ionotropic glutamate receptors (iGluRs) mediate fast synaptic transmission and are fundamental to processes such as long-term potentiation (LTP).
Mechanisms and Applications
Ionotropic receptors, including NMDA, AMPA, and kainate receptors, form ligand-gated ion channels that facilitate rapid changes in neuronal membrane potential. These receptors are crucial for synaptic plasticity—the cellular basis for learning and memory. Shah’s work underscores the importance of these receptors not only in normal cognitive function but also in the pathology of neurodegenerative diseases. Understanding the precise mechanisms of glutamate receptors could pave the way for new treatments that target cognitive deficits in conditions like Alzheimer’s disease.
Metabotropic Glutamate Receptors: Modulators of Synaptic Activity
While ionotropic glutamate receptors mediate fast excitatory signaling, metabotropic glutamate receptors (mGluRs) modulate synaptic transmission more slowly through G-protein-coupled mechanisms. For an in-depth look, refer to Metabotropic Glutamate Receptors.
Functional Insights
mGluRs are essential for fine-tuning neural communication, influencing synaptic plasticity, and modulating various intracellular signaling pathways. These receptors offer promising targets for therapeutic interventions in a range of neurological disorders, including chronic pain, anxiety, and depression. Shah’s research into mGluRs highlights how these receptors contribute to a balanced neural environment, providing a complementary perspective to the fast-acting ionotropic receptors.
Neurotransmitter Regulation and Signaling: The Master Switch
At the heart of neurotransmitter systems is the complex regulation of receptor activity and intracellular signaling pathways. In Neurotransmitter Regulation and Signaling, Nik Shah delves into the sophisticated mechanisms that control receptor function, including the roles of second messengers, receptor phosphorylation, and signal termination processes.
The Importance of Regulation
Proper regulation of neurotransmitter receptors ensures that signals are transmitted accurately and efficiently across synapses. Disruptions in these signaling pathways can lead to a host of neurological and psychiatric disorders. Shah’s insights into neurotransmitter regulation provide a comprehensive understanding of how receptor signaling is fine-tuned, opening up avenues for precise pharmacological interventions that target specific signaling cascades.
Opioid Receptors: Pain, Reward, and Beyond
Opioid receptors are pivotal in modulating pain, reward, and addictive behaviors. In Opioid Receptors, Shah examines the different subtypes of opioid receptors (mu, delta, and kappa) and their roles in mediating analgesia, mood, and stress responses.
Therapeutic Potential
The clinical implications of opioid receptor research are vast, especially in the context of pain management and addiction therapy. By understanding the precise functioning of these receptors, new drugs can be designed to minimize adverse effects such as tolerance and dependence. Shah’s work in this area underscores the potential for developing safer, more effective opioid therapies that target specific receptor subtypes.
Oxytocin and Endorphin Receptors: The Chemistry of Connection and Well-Being
Oxytocin and endorphins are often dubbed the “feel-good” chemicals of the brain. In Oxytocin and Endorphin Receptors, Nik Shah discusses how these receptors facilitate social bonding, emotional regulation, and pain relief.
Balancing Social and Emotional Health
Oxytocin is crucial for fostering trust, empathy, and social connections, while endorphins serve as natural pain relievers and mood enhancers. The intricate interplay between these receptor systems influences not only personal well-being but also broader social interactions. Shah’s research highlights that understanding these receptor mechanisms can lead to innovative treatments for conditions such as depression, anxiety, and social disorders.
Serotonin Receptor Families: Gatekeepers of Mood and Cognition
Serotonin is a key neurotransmitter involved in regulating mood, cognition, and a variety of physiological processes. In Serotonin Receptor Families, Shah provides a detailed analysis of the diverse serotonin receptors, including the 5-HT1, 5-HT2, 5-HT3, 5-HT4, 5-HT5, 5-HT6, and 5-HT7 families.
Comprehensive Overview
Each serotonin receptor family has distinct roles:
5-HT1 receptors generally mediate inhibitory effects and are linked to anxiety and depression.
5-HT2 receptors are involved in excitatory signaling and have implications in mood disorders and psychosis.
5-HT3 receptors function as ligand-gated ion channels, affecting rapid synaptic transmission.
5-HT4 receptors play a dual role in cognitive and gastrointestinal functions.
5-HT5, 5-HT6, and 5-HT7 receptors are emerging as key modulators of neuroplasticity, memory, and circadian rhythms.
Shah’s exploration of these receptor families provides a critical foundation for understanding how serotonin modulates both brain function and behavior. This knowledge is essential for developing targeted treatments for a range of mental health conditions.
Integrating Neurotransmitter Systems: A Holistic Approach to Brain Function
The study of neurotransmitter systems is not about isolated components; it is about understanding how these systems interact in a dynamic network to influence behavior, cognition, and overall health. Nik Shah’s work integrates multiple receptor systems to present a holistic view of brain function.
Cross-Talk Between Receptor Families
The interplay between dopamine, GABA, glutamate, opioid, oxytocin, endorphin, and serotonin receptors forms a complex network that underlies neural communication. For example, dopamine and serotonin systems often interact to balance mood and motivation, while GABA and glutamate receptors work in concert to regulate neuronal excitability and plasticity. Shah’s research in Neurotransmitter Regulation and Signaling emphasizes the importance of these interactions, revealing that disruptions in one system can have cascading effects on others.
Implications for Therapeutic Interventions
Understanding the integrated nature of neurotransmitter systems opens new avenues for therapeutic interventions. Targeted treatments that modulate multiple receptor systems simultaneously may prove more effective than those that focus on a single receptor type. Shah’s insights advocate for a multidimensional approach to pharmacotherapy—one that considers the broader context of neurotransmitter regulation and receptor cross-talk.
Practical Applications and Future Directions
Nik Shah’s extensive research into neurotransmitter systems provides a roadmap for future advances in neuroscience and mental health treatment. By leveraging insights from receptor biology, researchers can develop novel therapies for various neurological and psychiatric disorders.
Advancements in Precision Medicine
Personalized medicine is the future of healthcare. With a deeper understanding of neurotransmitter systems, clinicians can tailor treatments to an individual’s unique receptor profile. This precision approach has the potential to maximize therapeutic efficacy while minimizing adverse effects.
Innovative Drug Development
Shah’s work underscores the importance of developing drugs that target specific receptor subtypes. For instance, selective dopamine receptor modulators may offer new treatments for Parkinson’s disease and schizophrenia, while novel GABA receptor agonists could revolutionize the management of anxiety and epilepsy. Similarly, targeted therapies aimed at serotonin receptors hold promise for addressing mood disorders and cognitive impairments.
Integrative Therapeutic Approaches
Beyond pharmacology, integrating lifestyle interventions such as diet, exercise, and mindfulness practices can enhance neurotransmitter function and overall brain health. Shah’s research encourages a comprehensive approach that combines scientific advances with holistic wellness strategies—paving the way for a future where mental health is optimized through both medical and lifestyle interventions.
Conclusion: Embracing a New Era in Neurotransmitter Research with Nik Shah
The exploration of neurotransmitter systems and receptor families is at the heart of modern neuroscience. Nik Shah stands out as a pioneering figure whose work illuminates the intricate dance of dopamine, GABA, glutamate, opioid, oxytocin, endorphin, and serotonin receptors in regulating brain function and behavior.
By integrating insights from:
Dopamine Receptor Families
GABA Receptors and Their Subtypes
Glutamate and Ionotropic Receptors
Metabotropic Glutamate Receptors
Neurotransmitter Regulation and Signaling
Opioid Receptors
Oxytocin and Endorphin Receptors
Serotonin Receptor Families
—we gain a multi-layered understanding of how these receptor systems work individually and collectively. This comprehensive knowledge is crucial for advancing mental health research, designing innovative treatments, and ultimately enhancing brain function.
As the field of neuroscience moves toward a more integrated and precise understanding of neurotransmitter systems, Nik Shah’s visionary approach serves as both a beacon and a blueprint. By embracing his insights, researchers, clinicians, and health enthusiasts can look forward to a future where targeted, personalized interventions transform the landscape of mental health and cognitive well-being.
Embark on this journey into the intricate world of neurotransmitter systems with Nik Shah’s cutting-edge research as your guide. Discover how the modulation of receptor families can unlock new levels of brain performance and mental health, and join the movement toward a more nuanced, effective approach to neuroscience.
Explore More on @nikshahxai
Personal Development & Education
Philosophy, Ethics & Society
Technology & Innovation
Life Sciences & Health
About the Authors
For more information about Nik Shah's digital presence, as well as insights from contributing authors such as Nanthaphon Yingyongsuk, Sean Shah, Gulab Mirchandani, Darshan Shah, Kranti Shah, John DeMinico, Rajeev Chabria, Francis Wesley, Sony Shah, Dilip Mirchandani, Nattanai Yingyongsuk, Subun Yingyongsuk, Theeraphat Yingyongsuk, and Saksid Yingyongsuk, click here to explore further.
References
Nikshahxai. (n.d.). LinkTree Linktr.ee
#xai#nik shah#artificial intelligence#nikhil pankaj shah#nikhil shah#grok#claude#gemini#watson#chatgpt
1 note
·
View note
Text
a while ago i made a post outlining my experience of DXM significantly reducing my POTS symptoms. this happens consistently, whenever i have a flare up. since then i’ve looked into the mechanism of action of DXM and known biomarkers of POTS to see where interactions may occur. Overall, I think that dysregulation of glutamatergic signalling/excitotoxic damage and an increase in NADPH oxidase activity play a significant role in the pathophysiology of POTS. I made a post on neocities where I cover this in depth with references (linked above), but to summarise:
MRI studies have shown evidence of increased glutamate and excitotoxic damage in the brains of long covid patients as well as disrupted blood brain barrier integrity. a significant portion of long covid patients either have/develop POTS or experience orthostatic intolerance. from this you can infer that crossover is likely. as well as this, a range of biomarkers capable of contributing to glutamatergic dysregulation are confirmed in POTS patients. This includes enhanced platelet activation/aggregation, increased pro-inflammatory mediators, elevated growth hormone, decreased serotonin levels, autoantibodies against the nicotinic α3β4 and G-protein-coupled adrenergic A1 receptor. Increased plasma angiotensin II/angiotensin dysregulation has also been identified, which suggests increased activity of NADPH oxidase (leading to endothelial damage by reactive oxygen species).
DXM possesses several mechanisms capable of ameliorating this: • assists in regulating glutamate via suppressing overactivity of glutamatergic signalling as well as inducing uptake and recycling of glutamate from the extracellular space (reducing excitotoxicity) • directly targets NADPH Oxidase/NOX2, allowing for potent antioxidant + anti-inflammatory effects and vascular protection • a range of antiplatelet mechanisms • inhibition of serotonin reuptake
12 notes
·
View notes
Text
**Mechanism of Migraine with Aura: Step-by-Step Brain Tissue Changes**
Migraine with aura involves a cascade of neurological events, primarily driven by **cortical spreading depression (CSD)** and subsequent activation of pain pathways. Here’s a breakdown of the process:
---
### **1. Initiation of Cortical Spreading Depression (CSD)**
- **Trigger**: Neuronal hyperexcitability in the cortex (often genetic or due to ion channel dysfunction) leads to abnormal electrical activity.
- **Mechanism**: A sudden surge of neuronal depolarization (excessive firing) begins in a localized brain region, typically the **occipital lobe** (visual cortex).
- **Key Players**:
- **Glutamate**: Excess release of this excitatory neurotransmitter triggers depolarization.
- **Potassium (K⁺) ions**: Leak from neurons, propagating the wave.
---
### **2. Wave of Depolarization Spreads**
- **Process**: The depolarization wave spreads across the cortex at ~3–5 mm/minute, temporarily disrupting normal brain activity.
- **Symptoms**:
- **Visual aura**: Flashing lights, zigzag patterns (scintillations) or blind spots (scotoma) as the wave affects the visual cortex.
- **Sensory aura**: Tingling/numbness (parietal lobe involvement) or speech difficulties (Broca’s area).
---
### **3. Neurotransmitter & Ionic Shifts**
- **Glutamate Surge**: Sustained depolarization increases extracellular glutamate, overstimulating NMDA receptors and causing excitotoxicity.
- **Calcium Influx**: Neuronal calcium overload disrupts mitochondrial function, generating **reactive oxygen species (ROS)** and oxidative stress.
---
### **4. Cerebral Blood Flow Changes**
- **Initial Hyperemia**: Brief increase in blood flow (due to vasodilation) during depolarization.
- **Oligemia**: Prolonged reduced blood flow (vasoconstriction) following the wave, potentially contributing to hypoxia-like symptoms.
- **Imaging**: fMRI studies show a "spreading oligemia" matching the aura’s progression ([*Annals of Neurology*](https://doi.org/10.1002/ana.410280502)).
---
### **5. Trigeminovascular System Activation**
- **Trigeminal Nerve Stimulation**: CSD activates pain-sensitive trigeminal nerve terminals in the meninges (brain’s protective membranes).
- **Inflammatory Mediators**:
- **CGRP (Calcitonin Gene-Related Peptide)**: Released, causing vasodilation and neurogenic inflammation.
- **Substance P**: Enhances pain signaling and sensitizes blood vessels.
---
### **6. Central Sensitization & Pain**
- **Pain Pathways**: Trigeminal signals reach the thalamus and cortex, perceived as throbbing headache (often unilateral).
- **Sensitization**:
- **Allodynia**: Normal touch becomes painful (due to hyperexcitable neurons).
- **Photophobia/Phonophobia**: Brainstem nuclei (e.g., locus coeruleus) amplify sensitivity to light/sound.
---
### **7. Resolution & Postdrome**
- **Recovery**: Neurons repolarize, blood flow normalizes, and inflammatory mediators clear.
- **Postdrome**: Fatigue, cognitive fog, or residual sensitivity due to lingering neurotransmitter imbalances.
---
### **Key Factors Influencing Aura**
- **Genetics**: Mutations in ion channels (e.g., CACNA1A) increase neuronal excitability.
- **Hormones**: Estrogen fluctuations may modulate CSD susceptibility (common in women).
- **Triggers**: Stress, dehydration, or sleep changes lower the threshold for CSD.
---
### **Clinical Implications**
- **Preventive Therapies**: CGRP inhibitors, beta-blockers, or antiepileptics (e.g., topiramate) target CSD or trigeminal activation.
- **Acute Relief**: Triptans constrict dilated meningeal vessels and block CGRP release.
---
*Migraine with aura reflects a complex interplay of neuronal, vascular, and inflammatory processes. While CSD is central to aura, ongoing research aims to clarify genetic and molecular drivers.* 🧠⚡
0 notes
Text
oh also interesting to note: my theory of my issue primarily being glutamate excitotoxicity is further being supported by how my essential tremor was getting bad again in the two weeks i was without ketamine and now a day after a session my hands are rock steady. awesome
#excited to tell my psychatrist my theory and hear her thoughts on it#she's a researcher on ketamine therapy and is currently defending her thesis for her PHD abt it#go fuck em up girl
0 notes
Text
Part of the problem is that CFS/ME is unlikely to be just one pathology. Its a syndrome - a collection of symptoms. Thats part of why I have never been satisfied with that answer and so have tried to keep up with research into its pathology and comparing things I've found to my own symptoms. Following that research is what led to me reading about MTHFR mutations, which led me to taking methylfolate, which led to me discovering I likely have both a methylcobalamin deficiency and pyridoxal-5-phosphate deficiency that are the root of my CFS, POTS, worsening MCAS, hypersomina, and other progressive symptoms.
MTHFR mutations affect between 1 in 3 and 1 in 10 people, depending on race and nationality. They cause a deficiency in methylfolate which raises homocysteine levels and has a similar effect to a B12 or folate deficiency, but with normal B12 and folate levels.
Cobalamin metabolism disorders are more rare, but they are also very easily missed because total B12 blood tests are normal. The most common cobalamin metabolism disorder causes a deficiency of the usable forms of B12, leading to elevated methylmalonic acid and elevated homocysteine as would be seen in a B12 deficiency from diet or malabsorption. More rare disorders, or currently unknown and so undiagnosed disorders, can cause just a deficiency of methylcobalamin which will have nearly identical symptoms to a methylfolate deficiency.
One research team has hypothesized that an autoimmune disorder affecting the production of adenosylcobalamin may be behind multiple sclerosis. Impaired B12 metabolism leads to defective myelin, which the immune system tries to clear out. Something similar happens with methylcobalamin deficiency. Only genetic cobalamin metabolism disorders are recognized currently, but there could be autoimmune forms as well.
Similarly, some researchers think MTHFR mutations may be behind the trifecta of hEDS, POTS, and MCAS where patients often also fit CFS/ME and fibromyalgia criteria. Methylfolate deficiency can cause methylcobalamin deficiency, and methylcobalamin deficiency also leads to faulty myelin. Methylfolate and methylcobalamin deficiencies also cause problems with how DNA is read to make proteins, leading to faulty collagen, more damaged enzymes, and a variety of other metabolic problems.
The degree of impairment of the enzymes involved would impact the degree of severity of the damage done to the body. A mild deficiency over time would cause a slow, steady increase in platelet count, worsening allergies/MCAS reactions (homocysteine can trigger mast cells), and a wide variety of neurological symptoms as myelin degrades from lack of methylcobalamin and homocysteine causes excitotoxity.
While researching this, I found that there may also be millions of people with a functional pyridoxal phosphate deficiency who have no idea and no doctor is going to know to look for it. The people at risk are those who take B6 supplements and anyone who takes replacement thyroid hormones, both T4 and T3.
Most B6 supplements are a form of the vitamin called pyridoxine hydrochloride. This form can't be used by the body as is, it has to be converted into pyridoxal phosphate first. However, if taken at dosages that overwhelm the conversion to PLP, the pyridoxine itself can bind to PLP receptors on enzymes and block their function.
Free thyroid hormones also bind to PLP receptors, and they do it readily. A study in the 1950s found giving thyroid hormones to rats significantly blocked PLP enzyme function, especially in the liver. Ingested thyroid hormones are free when absorbed by the intestines and sent to the liver, and from there they can bind to carrier proteins which keep them from going places they aren't supposed to. In order to stop the thyroid hormones from interfering with the PLP enzymes, you need to significantly increase how much PLP is in the blood. How much more exactly is unknown, but is likely proportionate to the hormone dosage. Without supplementation or a diet quite high in natural B6 sources (not fortified sources), the body will start to use up the PLP it has stored up in the past. How fast someone depleates their PLP stores depends on how much B6 they get from their diet and thyroid hormone dosage. Functional PLP deficiency has the same symptoms as B6 deficiency, but bloodtests for the vitamin will be "normal." The demand for the vitamin is greater, so standard deficiency screenings aren't going to be useful.
PLP deficiency can also cause elevated homocysteine. Since enzymes in the intestines and liver are the most affected, people may have changes in gastric motility and in liver function tests. Over time, as the stores of PLP run low, people would develop signs of an overall B6 deficiency including neuropathy, dry and cracking skin, mood and sleep changes (PLP is necessary to make serotonin, dopamine, and melatonin), systemic inflammation and oxidative stress (PLP is needed to make the bodies most abundant antioxidant), and liver damage or disease.
Theres also a number of genetic conditions that can affect PLP levels, and, theoretically, there could be an autoimmune impact of PLP metabolism, so people who aren't taking thyroid hormones or pyridoxine can still have a PLP deficiency with normal total B6. PLP levels can be specifically tested and would show low in most of those cases.
The best news about both methylcobalamin and PLP deficiencies is that they are treatable without a prescription. Methylcobalamin lozenges or liquid for sublingual administration (to avoid the first pass effect which would make the supplement useless for the specific conditions that cause low methylcobalamin) and pyridoxal-5-phosphate/P5P/PLP are both available for a fairly low price online. Both are water soluble with limited uptake to storage and no toxicity, so whatever isn't either used or stored will be excreted in urine.
I have had CFS since fall of 2015. I was diagnosed with Hashimoto's and began taking thyroid hormones spring of that year. The onset and progression of all of my symptoms match having either a methylfolate or methylcobalamin deficiency combined with a PLP deficiency.
Taking methylfolate when you dont have a deficiency will increase demand for methylcobalamin, and if you can't meet that demand you will get sicker. Thats what happened to me, even though I was taking a normal B12 supplement and had high serum B12 - thus indicating the methylcobalamin deficiency. I got a methylmalonic acid blood test which indicated no deficiency of adenosylcobalamin, which eliminated the more common cobalamin metabolism disorders. I'd rather be extra rare than have a trio of deficiencies though.
If you take methylcobalamin without a deficiency, nothing changes and you pee it out. If you take it and you have a methylfolate deficiency, nothing changes and you pee it out. If you take pyridoxal phosphate without a deficiency, nothing changes and you pee it out.
These supplements do not interfere with any medications. Thyroid hormones can impair PLP enzymes, but PLP has no effect on thyroid hormone receptors.
This means that methylcobalamin and PLP are easily accessible and safe supplements that people with CFS/ME, fibromyalgia, and/or hEDS trifecta can try even if they don't have access to a helpful doctor.
The remaining question is how long they take to have effect. I can tell you that the PLP helped me in a matter of days. Better energy, better executive function, and some improvement to sleep. The methylcobalamin will take longer to have impact as the major effect of the deficiency is damage to the nervous system.
My neurological symptoms primarily come from the lumbar region of my spinal cord and the reticular formation of my brainstem. I dont know of any way to predict what parts of the nervous system get affected most, but by listing out all of my symptoms and learning the functions of different parts, I was able to trace all of my neurological symptoms to those two places.
Based on other types of B12 deficiency and other adult-onset cobalamin metabolism disorders, treatment has the potential to lead to full healing of damage done in the course of the deficiency. Lasting damage seemed to mostly be in cases where patients presented with strokes, embolism, etc. which caused additional damage. Other patients have full recovery as long as they continue to have the form of B12 they need.
I've so far only been taking the methylcobalamin for less than a week. Thats not enough time to recover from 8 years of damage, unfortunately. Right now I just have to wait and not push my neurological limitations just because I have more energy and focus. I have some very objective symptoms that are what I'll be monitoring most, since others like chronic widespread pain are more easily influenced by other factors. Im hoping that I will be able to point to definite improvement of at least a symptom after a month, but it could take longer. There's honestly no way to know.
Now, disclaimer, I am not a doctor or any other licensed medical professional. I'm a disabled person with the resources to experiment on myself and the ability and education necessary to understand the science enough to connect the dots and weigh the risks. I weighed the risk of taking methylfolate, but I turned out to be an outlier with a 1 in 100 million condition rather than the 1 in 3 for my demographic (granted the 1 in 100 million may be wrong, but thats the approximate current diagnosis rate). However, since what I found indicates zero risk for trialing methylcobalamin and/or PLP, I feel comfortable sharing what I've learned and what I am doing. Its always a good idea to talk to a pharmacist about possible interactions and to talk to a doctor about any other conditions you may have, and taking any supplement is always at your own risk.
I also understand the desperation of people chronic pain, fibromyalgia, and/or CFS/ME for anything that may help their condition, and it is hard for me to keep information to myself while I wait to see how things go, when the information has the potential to start helping someone who may not be able to make it until I have everything in a clear presentation format and my personal results.
As I continue my own treatment trial, I will be reaching out to various doctors and researchers about this and sharing more about my history, research, and how things are going. I will also be working on having cited, edited, clear, and easy to distribute information rather than... this or "look into trying these supplements and just trust me" which sounds about as useful and trustworthy as "try yoga."
even if you did the barest minimum research, the Wikipedia page for chronic fatigue syndrome is so sad. it’s like “this is what we diagnose people with when we can’t find anything else wrong with them. but it’s definitely something physical because there are visible neurological changes seen in neuroimaging and weirdness with the immune system. full recovery rates are less than 5%. the most common treatment is CBT which ignores the fact it is a physical illness and tells sick people they’ll get better if they just stop thinking about being sick.”
#medicine#science#chronic illness#chronic fatigue#cfs/me#rare disease#functional vitamin deficiency#long post
15K notes
·
View notes