#N501Y
Explore tagged Tumblr posts
Visit Tumblr Blog
Explore Tumblr blogs with no restrictions, modern design and the best experience.
Last Seen Tumblr Blogs





Fun Fact
Celebrities use Tumblr as well.
Text
Also preserved in our archive
Hey! Look! A great breakdown of that thing I'm always talking about being a big yet entirely-overlooked deal by 90% of medical professionals in regards to this particular virus!
SARS-CoV-2, the virus behind COVID-19, is not done with us. Over the past four years, it has shown a remarkable ability to adapt, with each new variant outmaneuvering our immune systems in unique ways. The recently published study on the XEC variant (November 22, 2024) provides fresh insights into how this virus is evolving. (1) Combining this with the broader history of immune evasion, we see a troubling pattern: the virus continues to find ways to evade the immune system and in many cases, persist, potentially leaving lasting impacts on our health even for those who experience only mild or asymptomatic infections.
What the Study Found: XEC’s Immune Escape Arsenal The latest study revealed that the XEC variant—an offspring of two previous variants, KS.1.1 and KP.3.3—has developed mechanisms that make it harder for our immune systems to neutralize it. Here’s how it works:
1. Glycosylation Mutations in the N-terminal Domain (NTD):
The XEC variant introduces new glycosylation sites, such as the T22N mutation, which act like a cloak, hiding key parts of the virus from antibodies.
These sugar molecules shield the receptor-binding domain (RBD), a crucial target for vaccines and natural immunity, making it harder for antibodies to bind and neutralize the virus.
2. Allosteric Effects:
Mutations in the NTD don’t just shield the virus—they also alter the behavior of the RBD through a process called allostery. These changes can make the RBD less accessible or alter how it interacts with human cells, further reducing the effectiveness of antibodies.
3. Potential Impact on Membrane Fusion:
The study hints that these mutations may also enhance how efficiently the virus fuses with human cells, potentially increasing its infectivity.
Immune Evasion: A Constant Tug-of-War The ability of SARS-CoV-2 to adapt is not new. Looking back at the history of immune evasion, we see a pattern:
The Early Days: Mutations like D614G made the virus more infectious.
Alpha and Beta Variants: N501Y and E484K mutations increased binding to human cells and evasion of neutralizing antibodies.
Omicron Era: A flurry of spike protein mutations allowed the virus to reinfect people with previous immunity and bypass vaccine-induced protection.
XEC is the next chapter in this story, combining these strategies with new tricks like glycosylation and allosteric modulation to stay ahead of human defenses.
Why This Matters: Beyond Infections Understanding immune evasion isn’t just about tracking infections—it’s about long-term health impacts. Here’s why this evolution is particularly concerning:
1. The Shadow of Long COVID:
Millions of people suffer from Long COVID, characterized by fatigue, brain fog, heart palpitations, and muscle pain. The virus’s ability to persist and evade the immune system might explain why symptoms linger for months or years in some individuals.
Chronic immune activation or hidden reservoirs of the virus could drive these long-term effects.
2. Asymptomatic but Chronic Damage:
Even in people without noticeable symptoms, SARS-CoV-2 has been shown to cause subtle, potentially long-term damage to:
Vascular systems: Leading to inflammation and microclot formation.
Neurological function: Disrupting brain activity and potentially accelerating neurodegenerative conditions. Early onset dementia
Musculoskeletal health: Causing unexplained weakness or pain.
Cognitive performance: Contributing to memory issues and reduced mental clarity. Are you or someone you know having more trouble finding words to use or losing things more often?
3. Vaccines Alone Aren’t Enough:
While vaccines remain essential, their effectiveness is limited by the virus’s rapid evolution. Variants like XEC show how SARS-CoV-2 can sidestep even the most advanced immune defenses, highlighting the need for next-generation vaccines targeting broader parts of the virus. We have know this for a long time now so where are the broader targeting vaccines?
The Future of SARS-CoV-2 Evolution The virus has already demonstrated its ability to adapt to our immune responses in multiple ways, and there’s no reason to believe it will stop. Here are some possibilities for future adaptation:
Further Refinement of Glycosylation: Adding or modifying sugar molecules could make the virus even more difficult to detect.
Enhanced Membrane Fusion: Mutations that improve how the virus merges with human cells could increase its infectivity.
Host Adaptation: Over time, the virus could become better at hiding within human cells, evading both natural immunity and therapeutic interventions.
Increased Chronicity: The virus might evolve to persist at low levels in the body, leading to ongoing inflammation and long-term health consequences.
What We Can Do: Adapting to the Virus’s Adaptations The XEC variant and others like it remind us that SARS-CoV-2 is still a formidable opponent. Here’s what we can do:
1. Invest in Better Vaccines:
Universal or pan-coronavirus vaccines that target conserved regions of the virus are critical.
2. Improve Diagnostics:
Detecting chronic or asymptomatic infections early could help mitigate long-term health effects.
3. Focus on Treatment:
Antiviral drugs that target different parts of the virus, combined with treatments for inflammation and immune dysregulation, could help reduce the impact of Long COVID.
4. Stay Vigilant:
For individuals, maintaining basic preventive measures during high transmission periods can significantly reduce risks.
Conclusion: Learning from the Virus SARS-CoV-2 is teaching us a harsh lesson about evolution. Its ability to adapt and evade our defenses, from antibodies to T-cells, shows no sign of slowing down. Variants like XEC underscore the importance of continued research, innovation, and public health vigilance. By understanding the virus’s strategies and preparing for its next moves, we can better protect ourselves—not just from acute infections but from the long-term consequences.
Reference:
Enhanced immune evasion of SARS-CoV-2 variants KP.3.1.1 and XEC through N-terminal domain mutations (November 22, 2024)
www.thelancet.com/journals/laninf/article/PIIS1473-3099%2824%2900738-2/fulltext
#mask up#public health#wear a mask#pandemic#covid#covid 19#wear a respirator#still coviding#coronavirus#sars cov 2
93 notes
·
View notes
Text
The Untold Story of SARS-CoV-2 Spike Variants Revealed: How Their Receptor Binding Domain Impacts Virus Interactions with Human Cells
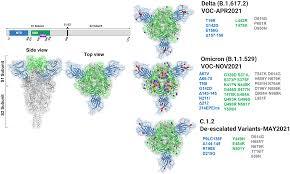
The Untold Story of SARS-CoV-2 Spike Variants Revealed: How Their Receptor Binding Domain Impacts Virus Interactions with Human Cells The Untold Story of SARS-CoV-2 Spike Variants Revealed: How Their Receptor Binding Domain Impacts Virus Interactions with Human Cells The spike protein of SARS-CoV-2 is a crucial determinant of infection and a key target for vaccines and therapeutics. Recent studies have identified several spike variants with mutations in the receptor binding domain (RBD) that are associated with changes in virus infectivity and immune evasion. This article aims to provide an overview of the untold story of SARS-CoV-2 spike variants and how their RBD impacts virus interactions with human cells. What is the RBD of SARS-CoV-2 Spike Protein? The spike protein of SARS-CoV-2 is a trimeric glycoprotein that mediates virus entry into host cells by binding to the ACE2 receptor on the cell surface. The RBD of the spike protein is a small domain that interacts specifically with the ACE2 receptor and is critical for virus attachment and entry. The RBD consists of a core and a receptor-binding motif (RBM), which is a flexible loop that undergoes conformational changes during ACE2 binding. What are the Spike Variants and their RBD Mutations? Several spike variants of SARS-CoV-2 have been identified, including the UK variant B.1.1.7, the South Africa variant B.1.351, the Brazil variant P.1, and the California variant B.1.427/B.1.429. These variants have mutations in the RBD that affect virus infectivity and immune evasion. For example, the UK variant has a mutation N501Y, which is thought to increase virus transmissibility by enhancing ACE2 binding. How Do Spike Variants Affect Virus Infectivity and Immune Evasion? Spike variants with RBD mutations can affect virus infectivity and immune evasion in several ways. First, mutations can increase or decrease ACE2 binding affinity, which affects virus entry into host cells. Second, mutations can alter the conformation of the RBD and affect antibody recognition and neutralization. Third, mutations can lead to the accumulation of multiple mutations in the RBD that enhance immune evasion and reduce vaccine efficacy. These effects have important implications for the development of vaccines and therapeutics against SARS-CoV-2. How Do Spike Variants Impact Vaccine Efficacy? Spike variants with RBD mutations can impact vaccine efficacy by reducing the neutralizing activity of antibodies generated by vaccination. For example, the South Africa variant B.1.351 has multiple mutations in the RBD that reduce the neutralizing activity of antibodies by up to 10-fold. This has prompted the development of new vaccine strategies that target multiple regions of the spike protein, rather than just the RBD. What Are the Implications of Spike Variants for Public Health? Spike variants of SARS-CoV-2 with RBD mutations have important implications for public health. These variants can increase virus transmissibility, reduce vaccine efficacy, and affect the severity of disease. To address these challenges, it is essential to continue monitoring SARS-CoV-2 variants and develop new vaccine and therapeutic strategies that are effective against multiple strains of the virus. How Does Research on Spike Variants Advance Our Understanding of SARS-CoV-2? Research on spike variants of SARS-CoV-2 advances our understanding of the virus by identifying key determinants of virus infectivity and immune evasion. This knowledge can inform the development of vaccines and therapeutics that are effective against multiple strains of the virus. Additionally, research on spike variants can help us understand the evolutionary dynamics of SARS-CoV-2 and the emergence of new variants in the future. What Are the Future Directions for Research on SARS-CoV-2 Spike Variants? Future research on SARS-CoV-2 spike variants should focus on several key areas. First, it is important to continue monitoring the emergence and spread of new variants and their impact on virus transmissibility, vaccine efficacy, and disease severity. Second, it is necessary to develop new vaccine and therapeutic strategies that are effective against multiple strains of the virus. Third, it is essential to understand the mechanisms of immune evasion and develop new strategies for overcoming this challenge. Conclusion In summary, the untold story of SARS-CoV-2 spike variants has revealed the importance of the RBD in virus infectivity and immune evasion. Spike variants with RBD mutations can impact virus transmissibility, reduce vaccine efficacy, and affect disease severity. To address these challenges, it is essential to continue monitoring variants and develop new vaccine and therapeutic strategies that are effective against multiple strains of the virus. FAQs: 1. How do RBD mutations affect SARS-CoV-2 infection? 2. What is the impact of spike variants on vaccine efficacy? 3. How does antibody recognition and neutralization play a role in RBD mutations? 4. Can vaccines be effective against multiple strains of SARS-CoV-2? 5. What are the future directions for research on SARS-CoV-2 spike variants? #TECH Read the full article
0 notes
Text
维州第二波疫情出现变异病毒 公众蒙在鼓里 | 病毒 | 病毒变种 | N501Y |
维州第二波疫情出现变异病毒 公众蒙在鼓里 | 病毒 | 病毒变种 | N501Y |
科学家认为,维州去年6月爆发的第二波病毒(COVID19)疫情,是目前具有高传染性的英国变种病毒的前驱病毒变异株所致,但当时政府并未告知公众,直至现在才被科学家曝出。 据悉尼晨锋报网站报导,墨尔本多尔蒂(Doherty)感染与免疫研究所分子病毒学实验室负责人珀塞尔(Damian…
View On WordPress
0 notes
Text
Pfizer’s vaccine can protect against coronavirus variant: Study
Pfizer’s vaccine can protect against coronavirus variant: Study
Image Source : AP Pfizer study suggests vaccine works against coronavirus variant New research suggests that Pfizer’s COVID-19 vaccine can protect against a mutation found in two highly contagious variants of the coronavirus that erupted in Britain and South Africa. Those variants are causing global concern. They both share a common mutation called N501Y, a slight alteration on one spot of the…
View On WordPress
#N501Y#Pfizer#Pfizer BioNTech#Pfizer BioNTech vaccine#Pfizer coronavirus vaccine#pfizer covid 19#pfizer covid-19 vaccine#Pfizer vaccine
0 notes
Text
New covid strain in England with 1000 confirmed cases. The mutations identified primarily affect the spike proteins, one mutation was the same one they saw in Denmark that let the virus jump to mink another is on the ACE-2 receptor and MAY increase infectivity and/or help the virus escape immunity gained from either having already had covid or being vaccinated.
They don't know if this new strain could allow for reinfection or not, but apparently these would be the mutations that would affect immunity response.
0 notes
Text
These new COVID-19 variants are causing global alarm beyond Delta strain
These new COVID-19 variants are causing global alarm beyond Delta strain
The continued spread of the SARS-CoV-2 virus has spawned a Greek alphabet of variants – a naming system used by the World Health Organization (WHO) to track concerning new mutations of the virus that causes COVID-19. Some have equipped the virus with better ways of infecting humans or evading vaccine protection. Scientists remain focussed on Delta, now the dominant variant around the world, but…
View On WordPress
#B.1.621 COVID-19 variant#COVID-19 variants#D614G COVID-19 variant#Delta covid-19 variant#E484K COVID-19 variant#lambda COVID-19 variant#mu COVID-19 variant#N501Y COVID-19 variant#SARS-CoV-2#SARS-CoV-2 virus#who#World Health Organization
0 notes
Text
How UK, South Africa Coronavirus Variants Escape Immunity
How UK, South Africa Coronavirus Variants Escape Immunity
All viruses mutate as they make copies of themselves to spread and thrive. SARS-CoV-2, the virus the causes COVID-19, is proving to be no different. There are currently more than 4,000 variants of COVID-19, which has already killed more than 2.7 million people worldwide during the pandemic. The UK variant, also known as B.1.1.7, was first detected in September 2020, and is now causing 98 percent…
View On WordPress
0 notes
Text
Inside the B.1.1.7 Coronavirus Variant
At the heart of each coronavirus is its genome, a twisted strand of nearly 30,000 “letters” of RNA. These genetic instructions force infected human cells to assemble up to 29 kinds of proteins that help the coronavirus multiply and spread.

As viruses replicate, small copying errors known as mutations naturally arise in their genomes. A lineage of coronaviruses will typically accumulate one or two random mutations each month.
Some mutations have no effect on the coronavirus proteins made by the infected cell. Other mutations might alter a protein’s shape by changing or deleting one of its amino acids, the building blocks that link together to form the protein.
Through the process of natural selection, neutral or slightly beneficial mutations may be passed down from generation to generation, while harmful mutations are more likely to die out.
Mutations In the B.1.1.7 Lineage
A coronavirus variant first reported in Britain has 17 recent mutations that change or delete amino acids in viral proteins.
The variant was named Variant of Concern 202012/01 by Public Health England, and is part of the B.1.1.7 lineage of coronaviruses.

Notable mutations in the B.1.1.7 lineage are listed below. Six other mutations, not shown in the diagram above, do not change an amino acid.
Eight Spike Mutations
Researchers are most concerned about the eight B.1.1.7 mutations that change the shape of the coronavirus spike, which the virus uses to attach to cells and slip inside.
Each spike is a group of three intertwined proteins:

Building one of these spike proteins typically takes 1,273 amino acids, which can be written as letters:

Spike proteins in the B.1.1.7 lineage have two deletions and six substitutions in this sequence of amino acids.

Written as letters, a B.1.1.7 spike protein looks like this:

These mutations alter the shape of the spike protein by changing how the amino acids fold together into a complex shape.
The Spike N501Y Mutation
Scientists suspect that one mutation, called N501Y, is very important in making B.1.1.7 coronaviruses more contagious. The mutation’s name refers to the nature of its change: the 501st amino acid in the spike protein switched from N (asparagine) to Y (tyrosine).

The N501Y mutation changes an amino acid near the top of each spike protein, where it makes contact with a special receptor on human cells.

Because spike proteins form sets of three, the mutation appears in three places on the spike tip:

In a typical coronavirus, the tip of the spike protein is like an ill-fitting puzzle piece. It can latch onto human cells, but the fit is so loose that the virus often falls away and fails to infect the cell.
The N501Y mutation seems to refine the shape of the puzzle piece, allowing a tighter fit and increasing the chance of a successful infection.

Researchers think the N501Y mutation has evolved independently in many different coronaviruses lineages. In addition to the B.1.1.7 lineage, it has been identified in variants from Australia, Brazil, Denmark, Japan, the Netherlands, South Africa, Wales, Illinois, Louisiana, Ohio and Texas.
In addition to N501Y, the B.1.1.7 has 16 other mutations that might benefit the virus in other ways. It’s also possible that they might be neutral mutations, which have no effect one way or the other. They may simply be passed down from generation to generation like old baggage. Scientists are running experiments to find out which is the case for each mutation.

One mysterious mutation in the B.1.1.7 lineage deletes the 69th and 70th amino acids in the spike protein. Experiments have shown that this deletion enables the coronavirus to infect cells more successfully. It’s possible that it changes the shape of the spike protein in a way that makes it harder for antibodies to attach.

Researchers call this a recurrent deletion region because the same part of the genome has been repeatedly deleted in different lineages of coronaviruses. The H69–V70 deletion also occurred in a variant that infected millions of mink in Denmark and other countries. Scientists are beginning to identify a number of these regions, which may play an important role in the virus’s future evolution.

In another recurrent deletion region, a number of coronavirus lineages are missing either the 144th or 145th amino acid in the spike protein. The name of the mutation comes from the two tyrosines (Y) that are normally in those positions in the protein.
Like the H69–V70 deletion, Y144/145 occurs on the edge of the spike tip. It may also make it harder for antibodies to stick to the coronavirus.


This mutation changes an amino acid from P to H on the stem of the coronavirus spike:

When spike proteins are assembled on the surface of a coronavirus, they’re not yet ready to attach to a cell. A human enzyme must first cut apart a section of the spike stem. The P681H mutation may make it easier for the enzyme to reach the site where it needs to make its cut.
Like N501Y, the P681H mutation has arisen in other coronavirus lineages besides B.1.1.7. But it’s rare for one lineage to carry both mutations.

ORF8 is a small protein whose function remains mysterious. In one experiment, scientists deleted the protein and found that the coronavirus could still spread. That suggests that ORF8 is not essential to replication, but it might still give some competitive edge over mutants that have lost the protein.
ORF8 is typically only 121 amino acids long:

But a B.1.1.7 mutation changes the 27th amino acid from Q to a genetic Stop sign:

Researchers assume that this ORF8 stump cannot function. But if losing the protein leaves B.1.1.7 at a disadvantage, it’s possible that the advantages of another mutation like N501Y might make up for the loss.

Detection and Spread
B.1.1.7 first came to light in the United Kingdom in late November. Researchers looked back at earlier samples and found that the first evidence dates back to Sept. 20, in a sample taken from a patient near London.
The B.1.1.7 lineage has now been detected in over 50 countries, including the United States. Britain has responded to the surge of B.1.1.7 with stringent lockdowns, and other countries have tried to prevent its spread with travel restrictions.

B.1.1.7 is estimated to be roughly 50 percent more transmissible than other variants. Federal health officials warn that it may become the dominant variant in the United States by March. It is no more deadly than other forms of the coronavirus. But because it can cause so many more infections, it may lead to many more deaths.

B.1.1.7 has been detected in at least 14 states, but the United States has no national surveillance program for determining the full extent of its spread.
How Did the Variant Evolve?
A number of researchers suspect that B.1.1.7 gained many of its mutations within a single person. People with weakened immune systems can remain infected with replicating coronaviruses for several months, allowing the virus to accumulate many extra mutations.
When these patients are treated with convalescent plasma, which contains coronavirus antibodies, natural selection may favor viruses with mutations that let them escape the attack. Once the B.1.1.7 lineage evolved its battery of mutations, it may have been able to spread faster from person to person.
Other Mutations in Circulation
One of the first mutations that raised concerns among scientists is known as D614G. It emerged in China early in the pandemic and may have helped the virus spread more easily. In many countries, the D614G lineage came to dominate the population of coronaviruses. B.1.1.7 descends from the D614G lineage.

A more recent variant detected in South Africa quickly spread to several other countries. It is known as 501Y.V2 and is part of the B.1.351 lineage. This variant has eight mutations that change amino acids in the spike protein. Among these mutations is N501Y, which helps the spike latch on more tightly to human cells.

None of these variants are expected to help the coronavirus evade the many coronavirus vaccines in clinical trials around the world. Antibodies generated by the Pfizer-BioNTech vaccine were able to lock on to coronavirus spikes that have the N501Y spike mutation, preventing the virus from infecting cells in the lab.
Experts stress that it would likely take many years, and many more mutations, for the virus to evolve enough to avoid current vaccines.
Sources: Andrew Rambaut et al., Virological; Andrew Ward, Scripps Research; Trevor Bedford, nextstrain.org; Paul Duprex, University of Pittsburgh School of Medicine; Houriiyah Tegally et al., medRxiv; Nature; Centers for Disease Control and Prevention; Global Report Investigating Novel Coronavirus Haplotypes. Spike models from Ward Lab, Scripps Research. Spike-receptor model by Cong Lab, Chinese Academy of Sciences. ORF8 model by the Yang Zhang Research Group, University of Michigan. Cahill-Keyes map projection by Gene Keyes. By Jonathan Corum and Carl Zimmer (The New York Times).
#science#medicine#virology#molecular biology#B.1.1.7#public health#COVID-19#academia#coronavirus mutations#infectious diseases#medblr
260 notes
·
View notes
Link
Serum samples from 20 individuals who received the Pfizer-BioNTech vaccine against SARS-CoV-2 thwarted a version of the coronavirus with the so-called N501Y mutation, according to a preprint posted to bioRxiv yesterday (January 7). This mutation is one of many sequence changes present in the B.1.1.7 and 501.V2 variants of SARS-CoV-2 that were first detected in the UK and South Africa, respectively, and are now rapidly spreading around the world.
“There’s no reason to think the vaccines won’t work just as well on these strains,” Frederic Bushman of the University of Pennsylvania who tracks how the virus mutates and was not involved in the work, tells the Associated Press. But he adds that the study only examined one mutation and the B.1.1.7 and 501.V2 variants have many more mutations that were not tested.
N501Y resides within the coronavirus’s spike protein that enables entry into host cells. Scientists at the University of Texas Medical Branch at Galveston had already engineered a version of SARS-CoV-2 with the N501Y mutation to study in mice when the new variants emerged, The Washington Post reports. The researchers collaborated with scientists at Pfizer to expose serum—an antibody-containing component of blood—from vaccine recipients to the engineered virus, and found no differences in neutralization between the N501Y virus and virus with the original Y501 sequence.
Continue Reading.
184 notes
·
View notes
Text
“But the concern is this virus is now radically different to the original that emerged in Wuhan, China. That means vaccines, which were designed using the original strain, may not be as effective.
Some of the mutations have been seen before in other variants, which gives some insight into their likely role in this variant.
For example N501Y seems to make it easier for a coronavirus to spread. There are some in there that make it harder for antibodies to recognise the virus and might make vaccines less effective, but there are others that are completely new.
Prof Richard Lessells, from the University of KwaZulu-Natal in South Africa, said: "They give us concern this virus might have enhanced transmissibility, enhanced ability to spread from person to person, but might also be able to get around parts of the immune system."”

Can’t fuckin’ do it, babes
8 notes
·
View notes
Photo
Mutation in SARS-CoV-2 Variant Does Not Affect Vaccine: Study
An engineered coronavirus with the N501Y mutation—one of many mutations present in the emerging B.1.1.7 and 501.V2 variants of the coronavirus—is neutralized by the sera of COVID-19 vaccine recipients.
Serum samples from 20 individuals who received the Pfizer-BioNTech vaccine against SARS-CoV-2 thwarted a version of the coronavirus with the so-called N501Y mutation, according to a preprint posted to bioRxiv yesterday (January 7). This mutation is one of many sequence changes present in the B.1.1.7 and 501.V2 variants of SARS-CoV-2 that were first detected in the UK and South Africa, respectively, and are now rapidly spreading around the world.
“There’s no reason to think the vaccines won’t work just as well on these strains,” Frederic Bushman of the University of Pennsylvania who tracks how the virus mutates and was not involved in the work, tells the Associated Press. But he adds that the study only examined one mutation and the B.1.1.7 and 501.V2 variants have many more mutations that were not tested.
N501Y resides within the coronavirus’s spike protein that enables entry into host cells. Scientists at the University of Texas Medical Branch at Galveston had already engineered a version of SARS-CoV-2 with the N501Y mutation to study in mice when the new variants emerged, The Washington Post reports. The researchers collaborated with scientists at Pfizer to expose serum—an antibody-containing component of blood—from vaccine recipients to the engineered virus, and found no differences in neutralization between the N501Y virus and virus with the original Y501 sequence.
According to Reuters, Pfizer had challenged its vaccine against 15 other mutations previously, finding them all to be inconsequential. “So we’ve now tested 16 different mutations, and none of them have really had any significant impact. That’s the good news,” Philip Dormitzer, Pfizer’s vice president and chief scientific officer of viral vaccines, tells Reuters. “That doesn’t mean that the 17th won’t.”
In particular, scientists have expressed concern about a mutation in 501.V2 called E484K, which is next to be tested, Dormitzer tells the AP.
Coauthor Pei-Yong Shi of UTMB tells the Post he expects to receive a viral variant next week to study in the lab. Moderna, AstraZeneca, and other vaccine makers are also in the process of challenging their vaccines with the B.1.1.7 and 501.V2 variants. Bushman tells the AP he expects similarly positive results. “A mutation will change one little place, but it’s not going to disrupt binding to all of them.”
Nevertheless, vaccine developers have not ruled out the possibility that a variant could evolve that would require reformulating vaccines. “These data don’t suggest a need for a change, but the mutations are hitting close enough to home that we need to be prepared,” Dormitzer tells STAT.
By Kerry Grens (The-Scientist). Image: A patient cell infected with SARS-CoV-2, NIAID
88 notes
·
View notes
Text
Omicron News Roundup
Reposting links from Naked Capitalism, a great blog that I can't recommend enough.
Link to the study itself:
To estimate the growth of the Omicron variant of concern (1) and its immune escape (2–9) characteristics, we analysed data from all PCR-confirmed SARS-CoV-2 cases in England excluding those with a history of recent international travel. We undertook separate analyses according to two case definitions. For the first definition, we included all cases with a definitive negative S-gene Target Failure (SGTF) result and specimen dates between 29/11/2021 and 11/12/2021 inclusive. For the second definition, we included cases with a positive genotype result and specimen date between 23/11/2021 and 11/12/2021 inclusive. We chose a later start date for the SGTF definition to ensure greater specificity of SGTF for Omicron.
We used logistic and Poisson regression to identify factors associated with testing positive for Omicron compared to non-Omicron (mostly Delta) cases. We explored the following predictors: day, region, symptomatic status, sex, ethnicity, age band and vaccination status. Our results suggest rapid growth of the frequency of the Omicron variant relative to Delta, with the exponential growth rate of its frequency estimated to be 0.34/day (95% CI: 0.33-0.35) [2.0 day doubling time] over the study period from both SGTF and genotype data. The distribution of Omicron by age, region and ethnicity currently differs markedly from Delta, with 18–29-year-olds, residents in the London region, and those of African ethnicity having significantly higher rates of infection with Omicron relative to Delta.
Hospitalisation and asymptomatic infection indicators were not significantly associated with Omicron infection, suggesting at most limited changes in severity compared with Delta.
To estimate the impact of Omicron on vaccine effectiveness (VE) for symptomatic infection we used conditional Poisson regression to estimate the hazard ratio of being an Omicron case (using SGTF definition) compared with Delta, restricting our analysis to symptomatic cases and matching by day, region, 10-year age band, sex and ethnicity. We found a significant increased risk of an Omicron case compared to Delta for those with vaccine status AZ 2+weeks post-dose 2 (PD2) , Pfizer 2+w PD2, AZ 2+w post-dose 3 (PD3) and PF 2+w PD3 vaccine states with hazard ratios of 1.86 (95%CI: 1.67-2.08), 2.68 (95%CI: 2.54-2.83), 4.32 (95%CI: 3.84-4.85) and 4.07 (95%CI: 3.66-4.51), respectively, where PD3 states are categorised by the dose 1/2 vaccine used. Depending on the Delta VE estimates used (10), these estimates translate into Omicron VE estimates of between 0% and 20% PD2 and between 55% and 80% PD3 against Omicron, consistent with other estimates (11). Similar estimates were obtained using genotype data, albeit with greater uncertainty.
To assess the impact of Omicron on reinfection rates we relied on genotype data, since SGTF is associated with a higher observed rate of reinfection, likely due to reinfections typically having higher Ct values than primary infections and therefore being subject to a higher rate of random PCR target failure. Controlling for vaccine status, age, sex, ethnicity, asymptomatic status, region and specimen date and using conditional Poisson regression to predict reinfection status, Omicron was associated with a 5.41 (95% CI: 4.87-6.00) fold higher risk of reinfection compared with Delta. This suggests relatively low remaining levels of immunity from prior infection.
The N501Y mutation in Omicron is universally observed to increase affinity roughly 6-fold, yet other mutations in key sites like K417N, Q493R, and G496S were shown by deep mutational scanning to decrease affinity. Increased affinity for the receptor may account, in part, for increased transmissibility, but that is clearly not the whole story as Omicron is much more transmissible than any previously isolates, including Alpha, Beta, Gamma, and Delta.
In the following weeks, an estimated 29,812 people would be hospitalized with COVID-19 and 3,876 would die every day on average, according to this projection.
"The most pessimistic scenarios are scary. And we need to sort of equip ourselves to make changes — change policies, encourage more cautionary behavior — if and when we start to see hospitalizations tick up in this country," Meyers says.
But Meyers stresses that the most dire scenarios assume the very worst, including that the U.S. takes no additional measures or behavior changes to slow the spread of the virus, such as more masking and social distancing.
6 notes
·
View notes
Text
New strain of the COVID-19 causing virus in Southern France.
Known as 'IHU', the B.1.640.2 variant has been reported by researchers at institute IHU Mediterranee Infection in at least 12 cases, and has been linked to travel to African country Cameroon.

The yet-to-be peer-reviewed study, posted on the preprint repository MedRxiv on December 29, revealed that IHU has 46 mutations and 37 deletions resulting in 30 amino acid substitutions and 12 deletions.
Amino acids are molecules that combine to form proteins.
Fourteen amino acid substitutions, including N501Y and E484K, and nine deletions are located in the spike protein.
2 notes
·
View notes
Link
Abstract
Associations between vaccine breakthrough cases and infection by SARS coronavirus 2 (SARS-CoV-2) variants have remained largely unexplored. Here we analyzed SARS-CoV-2 whole-genome sequences and viral loads from 1,373 persons with COVID-19 from the San Francisco Bay Area from February 1 to June 30, 2021, of which 125 (9.1%) were vaccine breakthrough infections. Fully vaccinated were more likely than unvaccinated persons to be infected by variants carrying mutations associated with decreased antibody neutralization (L452R, L452Q, E484K, and/or F490S) (78% versus 48%, p = 1.96e-08), but not by those associated with increased infectivity (L452R and/or N501Y) (85% versus 77%, p = 0.092). Differences in viral loads were non-significant between unvaccinated and fully vaccinated persons overall (p = 0.99) and according to lineage (p = 0.09 – 0.78). Viral loads were significantly higher in symptomatic as compared to asymptomatic vaccine breakthrough cases (p < 0.0001), and symptomatic vaccine breakthrough infections had similar viral loads to unvaccinated infections (p = 0.64). In 5 cases with available longitudinal samples for serologic analyses, vaccine breakthrough infections were found to be associated with low or undetectable neutralizing antibody levels attributable to immunocompromised state or infection by an antibody-resistant lineage. These findings suggest that vaccine breakthrough cases are preferentially caused by circulating antibody-resistant SARS-CoV-2 variants, and that symptomatic breakthrough infections may potentially transmit COVID-19 as efficiently as unvaccinated infections, regardless of the infecting lineage.
#covid-19#covid-19 vaccine#sars-cov-2#breakthrough#family medicine#viral escape#immune escape#selection pressure#print this off later
4 notes
·
View notes
Text
Genomic characterization of SARS-CoV-2 gamma variant in Brazil

[Image description: drawing of a masked person and coronaviruses in front of a Brazilian flag.]
During the second coronavirus disease 2019 (COVID-19) wave in Brazil, the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) P.1 (Gamma) lineage has accounted for most of the genomes sequenced. The Gamma variant is considered one of the most relevant variants of concern (VOC) globally. Within the spike protein of the Gamma variant, there are ten non-synonymous mutations (K417T, N501Y, and E484K), which include three which are situated in the receptor-binding domain (RBD).
A higher case fatality rate has been associated with the Gamma variant, and this trait may be related to its genetic background. It remains largely unknown how the unique set of approximately 35 amino acid substitutes originated that characterize this variant. Among all coronavirus types, reassortment of entire genome segments by “copy choice” recombinations is well described. Alternatively, within the citizens of northern Brazil, there is high seroprevalence of anti-SARS-CoV-2 antibodies, which may indicate that strong selective pressure was the cause for the new lineage.
In this study, a multi-national team of researchers describe the full-length SARS-CoV-2 genomes of 44 clinical samples from Amazonas, Brazil, sequenced and analyzed by their group, and compare them to previously described Gamma variant from Brazil and worldwide. Tests for phylogenomics, recombination, phylogenetic analyses of spike and non-structural proteins from open reading frame (ORF) 1a, and the detection of selective pressure acting on these sequences, were performed to gain an understanding of the evolutionary forces driving the Gamma variant emergence and evolution.
A preprint version of this study, which is yet to undergo peer review, is available medRxiv* server.
Continue reading.
2 notes
·
View notes
Note
Buongiorno Doc, non so se intaserò il tuo ask box con domande già fatte, ma cos'è questa variante nuova del covid che stanno trovando nei vari stati? Mi ricordo che una volta in una domanda parlasti di due tipi di mutazioni, una diciamo "leggera" (non so come definirla) e una catastrofica....
Si parlava dell’ANTIGENIC DRIFT e dell’ANTIGENIC SHIFT del virus influenzale...
... ma tranquilli tutti che qua non è una roba degna di panico ma solo di attento interesse.
Intanto cominciamo col dargli un nome:
VUI – 202012/01
che sta per
The first Variant Under Investigation in December 2020
anche se negli studi sparsi qua e là è molto più facile che la troviate come
Lineage B.1.1.7
Si tratta di un cluster epidemico che si è visto aver raccolto TRE mutazioni filogenetiche già precedentemente individuate separatemente:
La mutazione N501Y è uno dei sei residui di contatto chiave all'interno del dominio di legame del recettore (RBD) ed è stata identificata come una crescente affinità di legame con l'ACE2 umano e murino.
La delezione Spike 69-70del è stata descritta nel contesto dell'evasione della risposta immunitaria umana, ma si è anche verificata un certo numero di volte in associazione con altri cambiamenti RBD.
La mutazione P681H è immediatamente adiacente al sito di scissione della furina, una posizione ben nota per il suo significato biologico.
In parole povere si tratta sempre dello stesso identico virus ma con ADATTAMENTI funzionali a una migliore penetrazione dello stesso nelle cellule da infettare e una migliore difesa verso gli attacchi del nostro sistema immunitario.
Adesso esiste un Sars-CoV2 con fucili a munizioni perforanti e giubbotti antiproiettile.
È meno letale?
Sembra di sì ma c’era da spettarselo visto che tutti sanno che se macelli una vacca ci mangi un mese ma che se invece la mungi mangi formaggio per tutta la vita, quindi è successo che nei tentativi di mutazione sono sopravvissuti quei virus che facevano solo ammalare e spandere il contagio piuttosto che quelli che immobilizzavano e stecchivano il vettore in poco tempo.
E comunque sembra che questa nuova variante non riesca a evadere dalla risposta immunitaria ottenuta mediante vaccino.
SEMBRA... ma come sempre dobbiamo aspettare e lasciar studiare gli addetti ai lavori.
23 notes
·
View notes