#histiocytic lymphoma
Explore tagged Tumblr posts
Visit Tumblr Blog
Explore Tumblr blogs with no restrictions, modern design and the best experience.
Last Seen Tumblr Blogs





Fun Fact
Tumblr’s website traffic is steadily declining.
Text
Endoxan N 500 mg Injection is a prescription medication known as cytophosphane, and this medicine is classified as an alkylating agent. You can buy Endoxan N 500 mg Injection online at Buysm and get up to 75% off with home delivery. Check Endoxan N 500 mg Injection Price, Uses, Side Effects and Dosage.
#Endoxan N 500 mg Injection#Cadila Healthcare Ltd#malignant lymphomas#Hodgkin’s disease#lymphocytic lymphoma#mixed-cell type lymphoma#histiocytic lymphoma#Burkitt’s lymphoma#multiple myeloma#leukaemias#mycosis#fungicides#neuroblastoma#adenocarcinoma of the ovary#retinoblastoma#breast carcinoma
1 note
·
View note
Text
Note that the studies that were released by companies affiliated with polluters happened in 2019, during the trump administration.
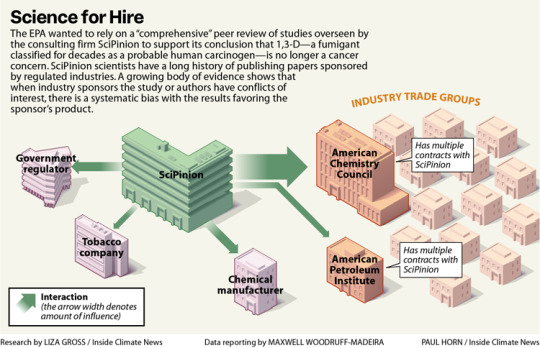
Excerpt from this story from Inside Climate News:
On a Southern California spring morning in 1973, a tanker truck driver jackknifed his rig and dumped the agricultural fumigant he was transporting onto a city street. A Los Angeles Fire Department emergency response team spent four hours cleaning up the chemical, 1,3-dichloropropene, or 1,3-D, a fumigant sold as Telone that farmers use to kill nematodes and other soil-dwelling organisms before planting.
Seven years after the spill, two emergency responders developed the same rare, aggressive blood cancer—histiocytic lymphoma—and died within two months of each other. In 1975, a farmer who’d accidentally exposed himself to 1,3-D repeatedly through a broken hose was diagnosed with another blood cancer, leukemia, and died the next year.
Within a decade of the men’s deaths, described as case studies in JAMA Internal Medicine, the National Toxicology Program, or NTP, reported “clear evidence” that 1,3-D causes cancer in both rats and mice. The finding led the U.S. Environmental Protection Agency to classify the chemical as “likely to be carcinogenic to humans” the same year, 1985. So it wasn’t a surprise when researchers at the University of California, Los Angeles reported in 2003 that Californians who’d lived at least two decades in areas with the highest applications of 1,3-D faced a heightened risk of dying from pancreatic cancer.
Yet EPA’s Office of Pesticide Programs’ Cancer Assessment Review Committee, or CARC, concluded in 2019 that 1,3-D—originally embraced by tobacco companies for its unparalleled ability to kill anything in soil that might harm their plants—isn’t likely to cause cancer after all.
In doing so, EPA, whose mission is to protect human health and the environment, rejected the human evidence, calling the UCLA study “low quality.” It also dismissed the authoritative NTP study and studies in lab animals that documented 1,3-D’s ability to damage DNA, a quintessential hallmark of cancer.
Instead, EPA’s CARC relied on studies provided by Dow AgroSciences (now called Corteva), the primary manufacturer of 1,3-D, and proposed a review of evidence linking the fumigant to cancer by SciPinion, a consulting firm hired by Dow, as an external peer review of its work. The decision to entrust external review to a Dow contractor has drawn repeated criticism, including from the agency’s watchdog, the Office of Inspector General, or OIG.
“During EPA’s search of the open literature, a comprehensive third-party peer review of the cancer weight-of-evidence assessment that considered toxicokinetics, genotoxicity and carcinogenicity data for 1,3-D was conducted and published in 2020 by SciPinion,” said agency spokesperson Timothy Carroll. EPA argued that the SciPinion review satisfied the criteria for an external review, Carroll said, and that another panel would have arrived at the same conclusion, given the specialized expertise required.
The OIG had recommended EPA conduct an external peer review of its 1,3-D cancer risk assessment in a 2022 report that outlined several problems with the agency’s process. An external review, the OIG said, requires “independence from the regulated business,” again noting the deficiency in a new report released in early August.
The scientists who run SciPinion have long consulted for manufacturers of harmful products, often publishing studies that deploy computer models to question the need for more protective health standards.
4 notes
·
View notes
Text
0 notes
Text
Reviewing questions:
Starting at age 30, FEV1 declines. Smoking causes COPD, which increases the rate of the decline. Smoking cessation slows the rate of decline in FEV1, but the gradual decline in FEV1 won't return to the way it would've been if the pt had never started smoking in the first place.
Bronchiolitis obliterans = obstruction of small airways (bronchioles); occurs in chronic lung transplant rejection. You get fibrosis of bronchioles.
Avitaminosis A (vitamin A deficiency)-> squamous metaplasia of the epithelium of the eyes, urinary, pancreatic, and respiratory tracts to a keratinizing epithelium. This can occur in pts with cystic fibrosis because the inspissated secretions block the pancreatic ducts, so you can't release the digestive enzymes necessary to absorb fat-soluble vitamins (A, D, E, and K).
Cigarette smoke leads to inflammation, leukocyte infiltration, protease-antiprotease (i.e., elastase-alpha 1 antitrypsin) imbalance, and oxidative stress, which lead to emphysema. Specifically, the smoke triggers alveolar macrophages to release TNF, which activates CD8+ T cells. Cells that cause inflammation (neutrophils) release elastase, which breaks down the elastin in the alveoli.
Aspiration pneumonia tx = clindamycin or beta-lactam and beta lactamase inhibitor.
Sarcoidosis = bilateral hilar lymphadenopathy; cough, night sweats. I remember learning in a lecture a long time ago that if the hilar LAD is not B/L, then you should be more worried about cancer. I thought this question was giving me B symptoms (weight loss, fever, night sweats), which are seen in B cell lymphoma (Hodgkin lymphoma) and it also mentioned supraclavicular nodes, which made me think cancer. The question also menitioned that the pt worked as a respiratory therapist, so I thought TB. Whatever. The answer was sarcoidosis. Non-caseating granulmonas are seen in sarcoidosis. It wasn't B cell lymphoma because B cell lymphoma would show Reed-Sternberg cells on biopsy, not granulomas.
In response to mycobacterium tuberculosis, Th1 cells relase interferon-gamma, which activates macrophages. Activated macrophages can then kill mycobacterium in phagolysosomes and become epithelioid and Langhans giant cells, which wall off the bacteria, forming granulomas.
Macrophages infected with M. tuberculosis present antigen to CD4+ Th0 cells. Macrophages also release IL-12 to Th0 cells. This triggers Th0 cells to become Th1 cells, which release interferon-gamma, which activates macrophages to kill M. tuberculosis intracellularly and to transform into epitheliod and Langhans giant cells to wall off the tuberculosis, creating granulomas. I've gotten questions on this several times now, so I better not forget it! Th1 cells also release TNF-alpha, which leads to more macrophages being recruited. You have to know what the epitheliod histiocytes and multinucleated giant cells look like on histology too.
Most cystic fibrosis pts die from cor pulmonale, bronchiectasis, or pneumonia. Makes sense since the inspissated secretions prevent proper alveolar ventilation-> pulmonary vasoconstriction-> pulmonary HTN.
The tendency of the lungs to collapse and the tendency of the chest wall to expand is balanced at functional residual capacity, which generates the negative intrapleural pressure that makes the chest wall and lung move together. A pneumothorax gets rid of the negative intrapleural pressure, and as a result, the chest wall expands to a new point of equilibrium. The hemithorax has a larger volume at the new equilibrium. The lung will collapse to the new equilibrium. Ok, I think I get it. The lungs want to collapse at all lung volumes. The chest wall wants to expand until large lung volumes. A PTX eliminates the intrapleural pressure that keeps the lungs and chest wall moving together, so then the chest wall expands. Compliance is change in volume/change in pressure. The pressure volume curve slope represents compliance; steep slop = high compliance. Hysteresis = lung compliance differs with inspiration and expiration because of alveolar surface tension. I remember one of the lecturers going over the pressure volume curves and he made a mistake or got confused trying to explain it. I think I get it.
Eosinophils have bi-lobed nuclei and granules that contain major basic protein. They attack parasites. Major basic protein can damage endo- and epithelial cells-> lung damage-> asthma.
If it's not strep pneumo, it's haemophilus influenzae or moraxella catarrhalis causing pneumonia. Infection with one of these bugs triggers COPD exacerbation (increased sputum production, dyspnea, cough, change in color of sputum). If it's viral, the most common cause is rhinovirus.
Sudden onset dyspnea = pulmonary embolism-> hypoxemia and hyperventilation. Hyperventilation causes respiratory alkalosis.
Small cell carcinoma is a neuroendocrine tumor (markers are Nueral Cell Adhesion Molecule [NCAM], neuron-specific enolase, chromagranin, and synaptophysin). Paraneoplastic syndromes are SIADH, Lambert-Eaton syndrome, and Cushing's syndrome. SIADH-> hypnatremia-> neuro symptoms. Histology shows sheets of small blue cells with little cytoplasm.
Once again, pulmonary arterial hypertension can be treated with endothelin receptor antagonists like bosentan. This prevents vasoconstriction and proliferation of vascular smooth muscle. In PAH, small arteries have proliferation of smooth muscle (hypertrophy of the tunica media) and fibrosis of the tunica intima (onion skinning). You also get capillary tufts, which are plexus-like lesions. The lumen of arteries decreases-> increased resistance-> pulmonary HTN. If it's familial PAH, it's due to inactivating mutations of BMPR2-> increased endothelin. The definitive treatment is lung transplant.
Intravenous drug abusers can get tricuspid valve endocarditis (using IV drugs-> introduction of staph aureus). The vegetations on the tricuspid valve can embolize and cause pulmonary infarcts, which are hemorrhagic. Specifically, these are septic pulmonary emboli, which cause wedge-shaped hemorrhagic infarcts in the lungs.
High altitude sickness can cause pulmonary edema (HAPE = High Altitude Pulmonary Edema) a few days after being at high altitude. Since there is less oxygen in the air at high altitudes, the less ventilated areas of lung have vasoconstriction, which is what the lungs do to divert blood flow to better ventilated areas of the lungs. But if enough of the lung vasoconstricts, then you get increased resistance to flow from the right heart to the pulmonary vasculature, so fluid backs up and can go into the lungs due to increased intravascular hydrostatic pressure. Pts present with dyspnea, cough, patchy alveolar infiltrates, crackles. They improve with supplemental oxygen. High altitude sickness doesn't just present as pulmonary edema. It can also cause acute mountain sickness (fatigue, nausea, HA) and cerebral edema (because decreased PaO2 causes increased cerebral flow; pts will be confused, lethargic, and have gait disturbance).
In COPD, FEV1 decreases more so than FVC, so the FEV1/FVC will be decreased. Reduced FEV1/FVC = obstructive lung disease.
A complication of pneumonia is an abscess, which occurs due to the release of granules by neutrophils. The granules kill bacteria, but also cause liquefactive necrosis of the lung tissue.
Electrical stimulation of the hypoglossal nerve (CN XII) can help improve obstructive sleep apnea, which I didn't know. But I guessed and got the question right. Stimulation of CN XII increases diameter of the oropharyngeal airway. Oh yeah, when I was listening to OnlineMedEd, Dustyn mentioned muscular weakness causing sleep apnea, like MS or something like that. So neuromuscular weakness can contribute to sleep apnea. The muscles relax when you sleep, so the airway can become obstructed.
When standing upright, ventilation is lowest at the apex of the lungs because gravity pulls the alveoli open, so when you inhale, the alveoli at the apex don't expand as much as the alveoli at the base of the lung. Basically, the alveoli at the apex already have the weight of the rest of the lung pulling the alveoli open, so they are already almost as expanded as they can get. The alveoli at the base of the lung don't have additional lung weight to pull them open, so they have more potential space to open. Thus, ventilation is greater at the base than the apex. When you inhale, since the alveoli at the base of the lung are not as expanded from the start of inhalation, they can expand more than the alveoli at the apex, thus they are better ventilated. I guess that's what this explanation was saying. But I remember learning that the apex is better ventilated whereas the base is better perfused. Idk. The base of the upright lung has better perfusion than the apex of the lung. The V/Q ratio is low at the base of the lung and high at the apex of the lung.
Theophylline is a bronchodilator that works like beta 2 agonists--it caueses increased cAMP-> smooth muscle relaxation. It is metabolized by the CYP450 system, so CYP450 inhibitors increase theophylline concentration-> theophylline toxicity (tremor, agitation, seizures, GI sxs, tachycardia, cardiac arrhythmias). Theophylline has a narrow therapeutic index and is not used much anymore. But it can be used for COPD or asthma. It's an adenosine receptor antagonist (similar to caffeine) and phosphodiesterase inhibitor. Infection with a fever can also cause theophylline toxicity.
Mucicarmine staining of BAL will show cryptococcus neoformans. The polysaccharide capsule of cryptococcus looks red with mucicarmine stain. India ink also shows the capsule. From Wikipedia: Mucicarmine stain is a staining procedure used for different purposes. In microbiology the stain aids in the identification of a variety of microorganisms based on whether or not the cell wall stains intensely red. Generally this is limited to microorganisms with a cell wall that is composed, at least in part, of a polysaccharide component. One of the organisms that is identified using this staining technique is Cryptococcus neoformans.
#aging#smoking#bronciolitis obliterans#sarcoidosis#tuberculosis#pneumonia#interferon gamma#IL 12#pneumothorax#PTX#pulmonary physiology#pulmonary#mountain sickness#high altitude sickness#theophylline#mucicarmine
4 notes
·
View notes
Text
Lupine Publishers | A Case Report of Tenosynovial Giant Cell Tumor in an Unusual Location: The Supraclavicular Region

Lupine Publishers | Journal of Otolaryngology
Abstract
Tenosynovial giant cell tumor (TSGCT) is a benign tumor that commonly presents in the upper limb joints and hands and less frequently occurs in the lower extremities. Usually they arise from synovium or tendon sheath. The aim of this article is to present an unusual location of TSGCT of the supraclavicular region in a young patient.
Keywords: Tenosynovial, giant cell tumor, supraclavicular region
Abbreviations: TSGCT: Tenosynovial Giant Cell Tumor, CSFIR: Colony Stimulating Factor 1 Receptor, WHO: World Health Organization
Introduction
Tenosynovial giant cell tumor (TSGCT) is a benign tumor that commonly presents in the upper limb joints and hands and less frequently occurs in the lower extremities. Usually they arise from synovium or tendon sheath [1,2] .TSGCT is divided into two main subtypes: the localized type that is otherwise known as giant cell tumor of tendon sheath that is well circumscribed and occurs in small joints e.g. fingers; it has a local recurrence rate of about (4- 30%) [1,2]. The second type is the diffuse TSCGT otherwise known as pigmented villonodular synovitis that is poorly circumscribed. It occurs usually in large joints and is more aggressive than type 1 TSCGT and has a recurrence rate of 18-50%. Type 2 TSCGT is cured with radical excision [2]. In this article, we report an unusual location of TSGCT of supraclavicular region, which is the second case that is presented in the literature [3].
Case Report
A 16-years old woman presented with a six-month history of slowly growing painless mass in left supraclavicular region. That was initially suspicious of a pathological lymph node, neurinoma or a hematological disorder (lymphoma). Neurological and vascular examination were unremarkable and laboratory values were within normal range. The patient underwent an ultrasound guided fine needle aspiration of the tumor that showed benign large multinucleated giant cells, histiocyte and xanthoma cells positive for CD68 by immunohistochemistry. The tumor showed no necrosis. The patient underwent further investigations including magnetic resonance imaging and angiography of neck (MRI and MRA scans). The results showed a 23x17x35 mm left supraclavicular mass that distorted scalene muscles in this region with normal vascular anatomy (Figure 1). The patient had an elective operation and the tumor was totally excised from level 5b left neck region and 2 lymph nodes were also removed (Figure 2). Postoperatively, the patient had an unremarkable recovery and she was discharged three days later. The histology confirmed the diagnosis of Tenosynovial giant cell tumor of left supraclavicular region. The lymph nodes showed no pathology.
Figure 1: Sagittal view MRI scan neck showing left posterior triangle mass.
Figure 2: Left supraclavicular tumor (bottom) and tissue with 2 lymph nodes(top).
Discussion
TSGCT or giant cell tumor of tendon sheath is a rare benign lesion involving joint synovia, bursae or tendon sheath. The annual incidence of TSGCT is 1.8 per million. It may be intra-articular or extra-articular and can be classified as local or diffuse lesions [1,4]. TSGCT is more common in women and the ratio is male to female 1:1.5 with average age between (30-50yrs) [1]. TSGCT may also affect children but the incidence for both localized and diffuse types is rare [5,6]. TSGCT is a monoarticular disease affecting typically large joints e.g. the knee. The second most affected joints are the hand and wrist followed by the hip and ankle. Common symptoms described in literature include pain, stiffness and swelling of the affected joint [7]. The tumor that we present is in an unusual site and according to literature is the second one described in the supraclavicular region [3]. Malignant tumors such as synovial sarcoma and benign tumors should be kept in mind as potential differential diagnosis [8,9]. Multinucleated giant cells are a major clue for diagnosis of TSGCT. On immunohistochemistry the histiocyte cells are positive for markers like CD68, CD45 and CD163 like it occurred in our case [10]. World Health Organization (WHO) classified TSGCT as a benign tumor. There is debate however whether it is a true neoplasm as studies have supported the presence of translocation in chromosome 1p11-13 and overexpression of macrophage colony stimulating factor 1 receptor (CSFIR) [7,11]. Initial treatment is surgical resection and if recurrence occurs then additional radiation therapy, synovectomy and external beam radiation therapy are recommended [12].
Conclusion
TSGCT is a benign tumor of tendon sheath of slow progression. As TSGCT in our case is located in an unusual location it is difficult to diagnose it. Diagnosis requires MRI scan and histologic confirmation. Initial treatment is surgical resection of the tumor. Recurrence rate does occur in TSGCT and therefore a close follow up is recommended.
Conflict of Interest
The authors have no conflict of interest to declare.
For more Otolaryngology Journals please click on below link https://lupinepublishers.com/otolaryngology-journal/
For more Lupine Publishers Please click on below link https://lupinepublishers.com/
#Lupine Publishers#Lupine Publishers Group#Scholarly Journal of Otolaryngology#Journal of Otolaryngology#SJO
0 notes
Text
The Monoclonal, Massive Globulin- Waldenstrom Macroglobulinaemia- Juniper Publishers

Preface
Wald Enstrom macrogobulinaemia is a disorder designated with a nomenclature of a Swedish physician Jan Gosta Waldenstrom (1906-1996). The exceptional disease was initially scripted in 1944 [1,2]. Waldenstrom macroglobulinaemia may be defined as the appearance of a serum para-protein such as immunoglobulin M (Ig M) in addition to a malignant lymphoplasmacytic infiltrate confined to the bone marrow. Lymphoplasmacytic Lymphoma (LPL) may be cogitated as a neoplasm comprising of miniature B lymphocytes, plasmacytoid lymphocytes and mature plasma cells. The tumefaction generally implicates the bone marrow with an occasional presence in the lymph node and spleen. Lymphoplasmacytic lymphoma is accompanied by Waldenstrom macroglobulinaemia in a majority (95%) of instances [1,2]. The dual conditions may be denominated by an immunoglobulin M (Ig M) monoclonal gammopathy accompanied by an emergence of a lymphoplasmacytoid lymphoma restricted to the bone marrow. Lymphoplasmacytoid lymphoma may concur with an infection of hepatitis C virus (HCV). A familial prevalence may be delineated. An estimated 1.4% of neoplasm of miniature B lymphocytes may be cogitated by lymphoplasmacytoid lymphoma [1,2].
Disease Characteristics
Lymphoplasmacytic lymphoma may be categorized as a post germinal centre B cell (CD10-, MUM 1+ and BCL6+/-) lymphoma commingled with divergent plasmacytic cellular differentiation. The lymphoma may depict concomitant infection with Hepatitis C Virus (HCV). The bone marrow infiltrate may predominantly be interstitial, nodular or of a diffuse configuration. Bone marrow trephine biopsy and bone marrow aspirate may demonstrate an admixture of miniature lymphocytes, plasmacytoid lymphocytes and mature plasma cells [2,3]. The malignant cellular egress may enunciate a monotypic secretion of serum immunoglobulin M (Ig M) protein which may be elucidated in a majority (> 90%) of instances. Subjects with Waldenstrom macroglobulinaemia frequently depict vascular hyper-viscosity. Thus, Waldenstrom macroglobulinaemia may be cogitated as a non-Hodgkin’s lymphoma concomitant with lymphoplasmacytic lymphoma asmajority (95%) of subjects of lymphoplasmacytic lymphoma elucidate features of Waldenstrom macroglobulinaemia. The indolent lymphoplasmacytic lymphoma and concomitant Waldenstrom macroglobulinaemia may exemplify a disorder of obscure origin [2,3]. Associated aspects of probable disease insurgence may be
1. Male sex
2. Enhancing age of disease emergence with a median age of diagnosis at 65 years,
3. A racial predisposition in Caucasians
4. The concurrence of immunoglobulin M monoclonal gammopathy of undetermined significance (Ig M MGUS).
Waldenstrom macroglobulinaemia may progress to adjunctive B lymphocyte malignancies with an estimated proportion of 10% at 5 years, 18% at 10 years and 24% at 15 years of disease incurrence with an overall ratio of malignant conversion at 1.5 % per year. The neoplasm also displays a familial preponderance and nearly 20% individual’s manifest family members suffering from Waldenstrom macroglobulinaemia and associated B lymphocyte malignancies. Environmental factors such as exposure to radiation or agent orange, hazardous occupation with handling leather, rubber, paints, dyes and solvents, coexistent autoimmune disease and infection with hepatitis C virus (HCV) may be incriminated in the evolution of the malignancy [3,4].
Clinical Elucidation
The circulation of serum monoclonal immunoglobulin M (Ig M) in Waldenstrom macroglobulinaemia may display characteristic constitutional symptoms with concurrent deposition of monoclonal immunoglobulin M (Ig M) protein in several body tissues with a consequent emergence of auto-antibodies. Waldenstrom macroglobulinaemia may manifest systemic symptoms with an estimated serum monoclonal immunoglobulin M (Ig M) protein greater than 3 grams/decilitre and a bone marrow ingress of malignant lymphoplasmacytoid cells greater than 20%. Approximately one fourth (27%) instances of Waldenstrom macroglobulinaemia may be asymptomatic, with anaemia in roughly 38% subjects, the emergence of hyper-viscosity in around 31% individuals, the appearance of B symptoms (fever, weight loss, night sweats) in nearly 23% and neurological symptoms in about 22% of patients [1,2]. Waldenstrom macroglobulinaemia may depict specific complications such as hyper-viscosity, tissue aggregation of immunoglobulin M (Ig M) or autoimmune haemolysis secondary to circulating macro-globulins. Subjects may present with haematemesis, haemorrhage from the nasal cavity and retinal vasculature [5,6]. Anaemia, thrombocytopenia, elevated Erythrocyte Sedimentation Rate (ESR), lymph node enlargement and hepato-splenomegaly may ensue. A bone marrow trephine biopsy may exemplify an abundance of malignant lymphoid cells. Radiographic analysis of the implicated bones may be unremarkable, thereby excluding a multiple myeloma. Serum protein examination may detect the presence of an extremely high molecular weight protein,” a macroglobulin”, cogitated as an excess of immunoglobulin M [5,6].The quantification of monoclonal immunoglobulin M (Ig M) may be concordant with the magnitude of bone marrow infiltration and severity of systemic symptoms. Hyper- viscosity may appear as chronic haemorrhage from the nasal cavity, gingiva or gastrointestinal tract accompanied by headache, dizziness, loss of coordination or balance, impaired hearing with tinnitus with blurring or loss of vision. Retinopathy may ensue on account of distended retinal veins and swelling of the optic disc. Severely affected subjects may display manifestations of heart failure, drowsiness, stupor and coma. Systemic symptoms may be discerned at a quantifiable serum Ig M value greater than 4000 milligrams/decilitre, though immunoglobulin levels may vary. Constitutional or B symptoms such as fever, weight loss greater than 10% of the body weight in preceding six months, drenching night sweats and fatigue may appear [6,7]. Peripheral neuropathy may be a manifestation of the disorder. Cold agglutinin disease may occur on account of elevated circulating antibodies to red blood cells which may aggregate at minimal body temperatures and induce a haemolytic anaemia along with Raynaud’s phenomenon, jaundice and haemoglobinuria. Cryoglobulinemia may be encountered with the precipitation of immunoglobulin M (Ig M) at reduced body temperatures in order to obstruct the miniature blood vessels with emerging consequences such as Raynaud’s phenomenon, thrombocytopenic purpura, haemorrhaging ulcers and gangrene of the fingers, toes, nose and ears [1,2].
Amyloidosis may occur with the configuration of an anomalous “amyloid” protein” which may accumulate in tissues and organs of the body such as gastro-intestinal tract, renal and hepatic tissue or heart and peripheral nerves. Malfunctions such as carpal tunnel syndrome, malabsorption, macroglossia, dermal thickening, swelling of the extremities, congestive heart failure and renal failure may emerge. “BING NEEL” syndrome may be cogitated with a lymphoplasmacytic infiltrate or deposition of immunoglobulin M (Ig M) within the central nervous system (brain or spinal cord). Systemic symptoms such as mental deterioration, confusion, visual disturbance, irritability, altered personality, convulsions and coma may concur. Recurrent sinus and upper respiratory tract infection, pleural effusion, pulmonary infiltrates and occasional rash may be delineated. Tumour cells of lymphoplasmacytic lymphoma may configure nodular aggregates in the skin, extremities, spine, breast and orbital socket [6,7].
Morphological Elucidation
Waldenstrom macroglobulinaemia with coexistent lymphoplasmacytic lymphoma enunciates malignant cells with characteristics of B lymphocytes and plasma cells, denominated as lymphoplasmacytic cells. A diffuse or interfollicular proliferation of malignant lymphoid cells may be cogitated. Cellular aggregates devoid of proliferation centres may be proportionately constituted by miniature B lymphocytes, plasmacytoid lymphocytes and mature plasma cells. A predominant lympho-plasmacytic infiltrate may be situated in the inter-trabecular region of the bone marrow [3]. Peripheral blood picture concordant with acute leukaemia may be demonstrated in an estimated one third (30%) instances. Tumour cells may predominantly omprise solely of miniature lymphocytes or small, mature lymphocytes commingled with plasmacytoid lymphocytes. The bone marrow may be infiltrated by an identical malignant infiltrate. Mature plasma cells, tissue mast cells and histiocytes may be quantifiably augmented. Plasma cells may infrequently be the preponderant cellular component. Serial sections from bone marrow trephine biopsy may delineate a diffuse or a focal lesion.The focal lesions may configure a para trabecular, interstitial or non paratrabecular pattern of tumour incrimination [3]. Expansive marrow replacement by the tumefaction may induce a significant reduction of normal haematopoiesis. Intra-nuclear inclusions termed as “Dutcher’ s bodies” may be articulated within lymphocytes and plasma cells and may be considered diagnostic of lymphoplasmacytic lymphoma. Intra-nuclear inclusions reactive to Periodic Acid Schiff’s (PAS) stain may similarly be configured in plasma cells constituting a multiple myeloma or reactive lymphoid proliferations. Lymphoplasmacytic lymphoma with Waldenstrom macroglobulinaemia may terminate with the evolution of a Richter’s syndrome, thereby recapitulating a Small Lymphocytic Lymphoma (SLL). The malignant egress may lack the presence of monocytoid cells, in contrast to a marginal zone lymphoma. The occurrence of Dutcher ‘s bodies with admixed enlarged, transformed lymphocytes may be characteristic of lymphoplasmacytic lymphoma. Mast cells may be intermingled with epitheloid histiocytes. Intercellular material stained with periodic acid Schiff (PAS+) stain may be exemplified along with scattered amyloid and crystal engulfing histiocytes [7,8].
Immune phenotype
Lymphoplasmacytic lymphoma cells may exemplify a surface immunoglobulin M (Ig M+). Immunoglobulin molecules confined to the cytoplasm may primarily be immunoglobulin M (Ig M) although Ig G or infrequently Ig A may be elucidated. The lymphoma may be immune reactive to CD20+ and associated pan B lymphocyte antigens such as CD19+, CD79a+ and PAX5+. However, a percentage may be non eactive for the aforementioned immune markers. Nonreactive immune molecules may be CD5-, CD10-, CD23- and BCL6- although CD5 may be debatable (-/+) (1,3). Plasmacytic immune markers CD38 and CD138 may be equivocal (+/-) in specific instances.
Molecular Characterization
A frequent genomic abnormality the MYD88L265P mutation may be discerned in a majority (95%) along with chromosomal deletion of del [6] (q21). Chromosomal translocation t (9:14) may be exceptional. The MYD88L265P chromosomal mutation may be universal in Waldenstrom macroglobulinaemia. A whole genome sequencing may depict the mutation in 90% instances. MYD88L265P chromosomal mutation may be infrequent in multiple myeloma, marginal zone lymphoma or immunoglobulin M –monoclonal gammopathy of undetermined significance (Ig M MGUS). Chromosomal mutation CXCR4 may be enunciated which may be identical to the WHIM syndrome (warts, hypogammaglobulinaemia, infections and myelokathexis). Individuals with Waldenstrom macroglobulinaemia devoid of MYD88 or a CXCR4 chromosomal mutation may depict an inferior survival, in contrast to instances delineating the mutations [1,2].
Differential Diagnosis
Lymphoplasmacytic lymphoma may necessitate a distinction from Chronic Lymphocytic Leukaemia (CLL), mantle cell lymphoma and plasmacytoid variants of extra nodal or nodal marginal zone lymphoma. Chronic Lymphocytic Leukaemia (CLL) may exhibit a focal plasmacytic differentiation. The tumour cells may be immune reactive to CD5+, CD23+ and a CD20 dim, in contrast to a lymphoplasmacytic lymphoma. Immunoglobulin M (Ig M) para-protein may be absent or minimal. Splenic marginal zone lymphoma may demonstrate an intra-sinusoidal pattern of marrow incrimination. Plasmacytic differentiation may be reduced or minimal. Immunoglobulin M (Ig M) para-protein may be lacking or be of miniscule quantities [1,2]. Distinction from plasmacytoid lymphoma may be particularly cogitated with demonstration of plasma cells and plasmacytoid lymphocytes, manifesting numerous inclusions confined to the cytoplasm which may react to the Periodic Acid Schiff ‘S (PAS+) stain. The tumour cells may thus recapitulate the appearance of histiocytes. Chromosomal point mutation MYD88 may be elucidated in a majority (90%) of instances of lymphoplasmacytic lymphoma, contrary to an exceptional delineation in multiple myeloma and marginal zone lymphoma [8,9] (Figures 1-14).
Criterion for Discerning Variants of Waldenstrom Macroglobulinaemia
1. A monoclonal gammopathy with Immune Globulin M (IgM) irrespective of the magnitude of M protein and an infiltration of lymphoplasmacytic cells greater than 10% in the bone marrow may be elucidated. Particularly inter-trabecular tumour dissemination may comprise of miniature lymphocytes with a plasmacytoid or plasma cell differentiation and a characteristic immune phenotype of immune reactive surface immune globulin M (Ig M+), CD19+, CD20+ and nonreactivity for CD5-, CD10-and CD23- . The aforementioned evaluation may competently exclude adjunctive lympho proliferative disorders such as Chronic Lymphocytic Leukaemia (CLL) and mantle cell lymphoma [1,2].
2. A monoclonal gammopathy of undetermined significance (Ig M MGUS) may enunciate serum monoclonal immunoglobulin M (Ig M) protein values below 3 grams/ decilitre with a lymphoplasmacytic cellular infiltrate beneath 10% generally confined to the bone marrow. Systemic symptoms of anaemia, hyper-viscosity, lymph node enlargement or hepato-splenomegaly may be absent. Immunoglobulin M monoclonal gammopathy of undetermined significance (Ig M MGUS) may evolve into a florid Waldenstrom macroglobulinaemia or an adjunctive B lymphocyte malignancy. The proportion of malignant transformation may emerge at an estimated 1.5% every year.
3. Smouldering Waldenstrom macroglobulinaemia may be an indolent or asymptomatic disorder. Monoclonal serum protein values for immunoglobulin M (Ig M) exceeding 3 grams/decilitre and/or a lymphoplasmacytic infiltrate restricted to the bone marrow in excess of 10% may be enunciated. End organ damage with coexistent anaemia, hyper-viscosity, lymph node enlargement or hepato-splenomegaly on account of a lymphoplasmacytic proliferation may be absent [1,2].
Investigative Assay
Discernment of Waldenstrom macroglobulinaemia mandates a disease evaluation with complete blood count, serum chemistries such as liver and renal function tests, blood glucose and specific procedures such as bone marrow trephine biopsy and bone marrow aspiration. Serum immunoglobulin assay may depict an overproduction of immunoglobulin M with a decline in the values of immunoglobulin G and A (Ig G and I gA), feature which may enhance the probability of emergent infections. Radiographic investigations may include a plain X-ray, Computerized Tomography (CT) scan, a Magnetic Resonance Imaging (MRI), an ultrasound and Positron Emission Tomography (PET) scan of the lymph node enlargement, enlarged spleen and dermal or tissue infiltrates of lymphoplasmacytic lymphoma cells [9,10]. Ocular examination may incorporate the assessment of retina and ocular fundus.
Therapeutic Options
Commencement of therapeutic intervention may concur with the appearance of B symptoms such as fever, night sweats, weight loss, fatigue, lymph node enlargement or splenomegaly. Haemoglobin declining to below 10 grams/decilitre or a platelet count below 100,000/ microlitre may be cogitated on account of bone marrow infiltration. Additionally, complications such as hyper viscosity syndrome, symptomatic sensory or motor peripheral neuropathy, systemic amyloidosis, renal insufficiency or symptomatic cryoglobulinaemia may require therapy [10,11]. A policy of careful observation may suffice for the indolent disorder. An estimated half (50%) of the subjects with Waldenstrom macroglobulinaemia devoid of constitutional symptoms and a lack of treatment at 3 years following diagnosis may be managed with “active surveillance “. Approximately 10% instances may not require a therapeutic intervention during a 10year duration following detection. It may be crucial to establish the emergence of a hyper-viscosity syndrome prior to initiation of therapy and if plasmapheresis may be necessitated. Waldenstrom macroglobulinemia when associated with hyper-viscosity may display systemic symptoms such as visual deterioration, neurological symptoms and haemorrhage, incurring with immunoglobulin M serum values greater than 4 grams/decilitre and the condition may be benefitted with plasmapheresis [11,12]. Chemotherapeutic agents found efficacious in Waldenstrom macroglobulinaemia include chlorambucil, cyclophosphamide, bendamustine, nucleoside analogues such as fludarabine and cladribine. Corticosteroids prednisone or dexamethasone may be applicable. Biologic therapy may enunciate the utilization of anti monoclonal antibody conjugates such as rituximab, ofatumumab or obinutuzumab. Immune modulators such as thalidomide and lenalidomide may prove to be effective [1,2]. Administration of proteasome inhibitors such as bortezomib, carfilzomib and ixazomib may be advantageous. Targeted therapy implicating the B cell signalling pathway may concur as imbruvica and everolimus [12,13]. Initial or preliminary therapy for previously untreated, symptomatic subjects may employ
a. purine analogues with a combination of fludarabine, cyclophosphamide and rituximab (FCR) or fludarabine and rituximab (FR).
b. Alkylating agents in varying combinations such as rituximab with cyclophosphamide, doxorubicin, vincristine and prednisone (R CHOP), dexamethasone, rituximab and cyclophosphamide (DRC) or rituximab with bendamustine (BR) may be beneficial
c. Bortezomib in diverse combinations such as bortezomib, dexamethasone and rituximab (BDR) may be applicable.
d. Singular antibody conjugate such as rituximab may be utilized for initiation of therapy.
e. Ibrutinib as a Bruton’s tyrosine kinase inhibitor (BTK inhibitor) may be efficacious in instances with MYD88 mutation. Concomitant chemo immunotherapy may be administered
Administration of bendamustine with rituximab may depict a median and inter-quartile range (IQR) of 69.5 months in patients of Waldenstrom macroglobulinaemia [1,2]. The application of R CHOP may display a median and inter-quartile range (IQR) of 28.1 months. Solitary administration of rituximab in Waldenstrom macroglobulinemia may exhibit an objective response rate (ORR) of 52%, a partial response (PR) of 27% and a minor response (MR) of 25%. The median duration of response (DOR) may be elucidated at 27 months. An estimated half (54%) of the individuals may depict an elevated immunoglobulin M (Ig M) “flare” and one fourth (27%) subjects may delineate a persistent elevation of serum immunoglobulin M at 4 months duration following discernment of disease. Administration of ibrutinib may demonstrate an objective response rate (ORR) of 90.5%, a partial response (PR) of 73% and the median time to suitable therapeutic response may appear at 4 weeks. The progression free survival (PFS) at the end of 2 years may be 69.1% and Overall Survival (OS) at 95.2 %. Toxicity levels to the chemotherapeutic agent may be greater than grade 2. Ibrutinib administration may be accompanied by specific toxicities such as thrombocytopenia, neutropenia, stomatitis, atrial fibrillation, diarrhoea, herpes zoster infection, haematoma, secondary hypertension and epistaxis [13].
Contemporary Instances
Contemporary instances of Waldenstrom macroglobulinaemia may necessitate management as described:
1. Monoclonal gammopathy of undetermined significance (Ig M MGUS) with a lymphoplasmacytic infiltrate below 10%, an asymptomatic or smouldering Waldenstrom macroglobulinaemia with haemoglobin greater than 11 grams/decilitre, a platelet count in excess of 120,000/ millilitre may be managed by” watchful waiting”.
2. A symptomatic Waldenstrom macroglobulinaemia with a haemoglobin level below 11 grams/decilitre or a platelet count beneath 120,000/ millilitre.
a. Immunoglobulin M (Ig M) related neuropathy.
b. Haemolytic anaemia secondary to Waldenstrom macroglobulinaemia.
c. Symptomatic cryoglobulinaemia. The aforementioned instances may be managed with a solitary antibody conjugate such as rituximab. A maintenance therapy may not be required. Plasmapheresis may be opted for in instances of hyper-viscosity secondary to chemotherapy [1,2].
3. Waldenstrom macroglobulinaemia elucidating bulky disease (tumour magnitude greater than 10 centimetres) or profound pancytopenia with reduced blood counts such as haemoglobin below 10 grams/decilitre or a platelet count beneath 100,000/ millilitre with the appearance of constitutional symptoms and a lack of features of hyper-viscosity or hyper-viscosity may be managed with plasmapheresis.
The aforementioned subjects may be administered concomitant bendamustine with rituximab with the regimen devoid of maintenance therapy with singular rituximab. Stem cell transplantation may be a pre-requisite. Alternatively, stem cells may be mobilized and cryo-preserved for subjects beneath ≤ 60 years of age or emerge as potential and future candidates of Autologous Stem Cell Transplantation (ASCT). Stem cell transplantation may be suitably employed with subjects of relapsed or refractory Waldenstrom macroglobulinaemia. Autologous stem cell transplant (ASCT) may depict a 5year Progression Free Survival (PFS) of 40% and an overall survival (OS) of 68%. Allogeneic stem cell transplant may exhibit a 5year progression free survival (PFS) of 56% and a 5year overall survival (OS) of 62% [1,2]. Clinical trials with novel agents or drug conjugates may be mandated. Contemporary drugs such as ibrutinib, a Bruton’s tyrosine kinase inhibitor or idelalisib, a PI3kinase inhibitor or everolimus, an m TOR inhibitor may be efficaciously adminstered. Contemporary anti CD20 antibody conjugates such as of tamumab, anti BCL2 agents such as venetoclax, recent histone deacetylase (HDAC) inhibitors such as panobinostat, recent proteasome inhibitor carfilzomib, recent immunomodulatory agents such as pomalidomide may be additionally assessed. Contemporary targeted therapies may include molecules such as ventoclax, acalabrutinib and BGB3111. The aforementioned drugs may be utilized in combination with established agents.
Salvage Therapy
Salvage Therapy may be applicable in specific instances. In subjects where the requirement of subsequent therapy may exceed 4 years, the original therapeutic regimen may be replicated. For therapeutic installation within 4 years, a monotherapy with ibrutinib or combinations such as Dexamathasone, Rituximab and Cyclophosphamide (DRC) may be opted for. Concomitant administration of bendamustine with rituximab (BR), Bortezomib, Dexamethasone and Rituximab (BDR) may be effective.
Supportive Therapy
Supportive Therapy may incorporate modalities such as blood transfusion, administration of growth factors in order to enhance the blood cell counts (white and red blood cells with platelets). Surgical procedures may be specified in particular instances in the form of splenectomy or plasmapheresis in order to reduce the serum immunoglobulin M (Ig M) quantities. Targeted radiation may be employed in order to decimate the magnitude of incriminated lymph nodes [12,13] Table 1.
Table 1: Distinction betwixt Lymphoplasmacytic Lymphoma (LPL) and small cell Plasma Cell Myeloma (PCM) [1,2].
https://juniperpublishers.com/ijcsmb/IJCSMB.MS.ID.555672.php
0 notes
Note
If boxers aren't in line to get evaluated, I'd like to put them there... No hurry. :)
Ah, Boxers. Clowning cancer factories. They’re such an interesting breed and frequent visitors to the vet clinic. They’re also one of the addictive breeds, meaning that despite their flaws there are a lot of people that once they own one, are never without one ever again. You might want to sit down and have a cup of tea.
Disclaimer: These posts are about the breed from a veterinary viewpoint as seen in clinical practice, i.e. the problems we are faced with. It’s not the be-all and end-all of the breed and is not to make a judgement about whether the breed is right for you. If you are asking for an opinion about these animals in a veterinary setting, that is what you will get. It’s not going to be all sunshine and cupcakes, and is not intended as a personal insult against your favorite breed. This is general advice for what is common, often with a scientific consensus but sometimes based on personal experiences, and is not a guarantee of what your dog is going to encounter in their life.

So, the number one thing that Boxers as a breed are known for in veterinary medicine, if there one one solitary defining feature that was the reason most veterinary professionals decide against owning a boxer, a breed they would otherwise like, then at the risk of being insensitive, (since you like sparkly gifs) its...

Boxers are prone to cancer like no other breed I know, closely followed by Golden Retrievers. They develop all sorts with great ease, at unfortunately young ages with great regularity.
Mast Cell Tumors are the bane of the boxer breed. These tumors can develop anywhere on the body, including in organs like the spleen, and in any layer of the skin. These tumors are sometimes called the Great Pretenders because they can look like lots of different things. They're easily mistaken for benign lipomas by feel, and can be misdiagnosed if they're growing under a lipoma by FNA as it's easy to miss a small lump with a small needle.While a low grade MCT has a chance to be cured with surgery of detected early, a high grade one is all kinds of trouble even with modern chemotherapy options. It's fear of these tumors that cause many vets, including myself, to be highly suspicious of every single lump on a boxer or boxer cross.Boxers also seem highly prone to other cancers too, lymphoma being high on the list. Individuals with a white belly also get squamous cell carcinomas and cutaneous haemangiomas.
They are one of the very few breeds known to develop malignant histiocytomas, which is especially unfortunate considering that in most dogs a histiocytoma goes away all on its own in a few months, but in Boxers it will potentially kill them.
So while any lump on any dog can be a malignant cancer, Boxer’s have the added ‘fun’ of developing lumps that probably would have been fine on an other dog and look benign but sometimes actually aren’t. Can you understand my paranoia?
Boxers are a brachycephalic breed, meaning they have shortened muzzles and flattened faces. There is significant individual variation within this breed, but more extreme individuals do suffer from Brachycephalic Airway Syndrome (BAS)
Their facial conformation leaves their eyes prone to numerous Eye Conditions, including but not limited to cherry eye, entropion, exposure keratopathy and corneal ulcers. They also get a particularly difficult to treat eye ulcer called ‘indolent ulcers’ which are sometimes just called ‘Boxer dog ulcers’. They also get progressive retinal atrophy which is probably more genetic than anything else.
Speaking of diseases that are names after the breed (rarely a good sign), this breed also gets an unusual gastrointestinal disease called Histiocytic Ulcerative Collitis, which is also called Boxer Dog Collitis. For brevity’s sake, think of it a bit like a type of IBD of Chron’s disease.
And while we’re still on the topic of diseases named after this breed, Boxer Cardiomyopathy, which is really a arrhythmogenic right ventricular cardiomyopathy that’s primarily identified in boxers, also afflicts this breed. It’s not their only heart condition though, Dilated cardiomyopathy, atrial-septal defect, subaortic stenosis and sick sinus syndrome also occur.
This is turning into a long post, isn’t it. Do you want a break? How about another gif?

Okay, let’s talk some more about Boxers from a veterinary standpoint.
Boxers are prone to a couple of neurological disorders, Wobbler Syndrome is more common in larger males but degenerative myelopathy can occur in any boxer, is they live long enough to get it.
Younger boxers may develop demodex, if they’re juvenile when they do so it’s likely due to a funky immune system, which might explain a lot about this breed. Boxers that are predominantly white may also be deaf in one of both ears. It’s claimed that white boxers are more prone to cancer too, and for skin cancers this is true, but all boxers are prone to cancer. Hence the sparkly gif.
Possibly related to an interesting immune system, the breed is prone to allergies and atopy. This is a day to day annoyance on top of he life threatening/shortening conditions this breed is likely to develop.
Speaking of life threatening, the boxer dog is certainly deep chested enough to develop Gastric Dilatation Volvulus and need a trip to the emergency clinic.
And possibly the least interesting thing on this list the breed is seen relatively frequently for in the veterinary clinic is hip dysplasia. Gosh, a long list never looks good, especially when three conditions are named after the breed.
Boxer’s also have a reputation for anaesthetic sensitivity. This is often exaggerated in breed circles, assuming the boxer in question doesn’t have one of the aforementioned heart conditions, but because they are brachephalic they have a higher vagal tone and are more sensitive to the common sedative acepromazine.
This doesn’t mean you can’t use acepromazine in boxers, only that you have to be careful with it. I will often use it at a tenth to a quarter the dose in young, nutty individuals before surgery, but some vets wont use it at all.
Can you see how living with one of these dogs would drive me nuts from a medical paranoia standpoint?
213 notes
·
View notes
Text
Biomed Grid | A Case of KiKuchi-Fujimoto Disease/Histiocytic Necrotizing Lymphadenitis in 25 years old African American Female
Introduction
Kikuchi-Fujimoto disease, also known as histiocytic necrotizing lymphadenitis, was first simultaneously described in 1972 by Kikuchi and Fujimoto [1,2]. Kikuchi disease has been described in both genders. Overall, there is a higher incidence in female patients. The reported age range of Kikuchi-Fujimoto disease is from 6 to 80 years with majority of the patients under the age of 40 years. The mean age at presentation in United States is 30 years of age. Even though this condition was originally reported mainly in young Japanese females, the ethnic distribution of Kikuchi disease in United States include a variety of ethnic background [3-5]. Despite its benign lymphadenopathy and self-limited clinical course, Kikuchi-Fujimoto disease requires vigilant examination in order to exclude conditions with overlapping clinical and histological features that necessitates different and more aggressive therapy.
Case Report
25 years old African American female present with 1-month duration of daily recurrent episodes of pyrexia (102 F) with painful left axilla lump. The left axilla lump was soft, tender, and mobile. Pa tient denies having fatigue, night sweats, shortness of breath, chest pain, nausea, vomiting, and diarrhea. Patient has had a decrease in appetite during the month which has led to a 10 lbs. weight loss. Patient felt dizzy at times and experienced loss of consciousness of short duration. Patient has also experienced a few days of dry cough.
Patient was prescribed Sulfamethoxazole/Trimethoprim and Cephalexin for a week. Shortly after, patient developed a rash. Drug allergy was suspected. Thus, patient was switched to clindamycin for 7 days. Without improvement, patient was sent to emergency department and had a septic workup with negative results for sepsis. Chest x-ray results were also negative
Subsequent left axilla lymph node biopsy was performed due to suspicion of lymphoma. Gross examination of the left axillary lymph node biopsy demonstrated fragments of tan white tissue measuring 0.3 x 0.2 x 0.1 cm in dimension
Microscopic examination of the ultrasound guided needle biopsy specimen demonstrates lymph node tissue with preserved follicular pattern and area of necrosis. Further immunohistochemical stains demonstrate CD20 and CD45 positive B-cells in follicular pattern; CD3, CD5 and BCL-2 positive T-cells in parafollicular pattern; scattered CD138 positive plasma cells; and some CD68 positive histiocytes (Figure 1). KI-67 labeling index is approximately 20%. CD10, CD15, CD30, and AE 1/3 had negative immunohistochemical staining result.
Figure 1:Immunohistochemistry demonstrating CD68 positive histiocytes.
The gender, age, symptoms, histological characteristics, and immunohistochemical profile supports the diagnosis of Kikuchi-Fujimoto disease.
Discussion
Low grade fever and painless moderate sized cervical lymphadenopathy in a previously well young woman is the most common clinical manifestation of Kikuchi-Fujimoto disease [3,6]. According to Bosch et al, patients may also experience painful lymphadenopathy. The affected lymph node size can vary from 0.5-4 cm with rare occurrence of enlarged lymph nodes greater than 6cm. 30% to 50% of patients with Kikuchi disease can present with symptoms of upper respiratory infection, weight loss, nausea, vomiting, sore throat, and night sweats [7]. According to the retrospective literature review by Kucukardali et al. on 244 cases of Kikuchi-Fujimoto disease, the most common signs and symptoms are and not limited to: lymphadenopathy (100 %), fever (35%), rash (10%), arthritis (7%), fatigue (7%), hepatosplenomegaly (3%) [8].
Due to unspecific laboratory and imaging characteristics, definitive diagnosis of Kikuchi-Fujimoto disease is centered on excisional lymph node biopsy and subsequent histopathological assessment [9,10]. Despite the self-limited nature of this syndrome, excisional lymph node biopsy is necessary in order to exclude clinically similar conditions requiring further aggressive therapy [7].
In terms of histology, the affected lymph nodes demonstrate focal, well-circumscribed, paracortical necrotizing changes. High power view of the necrotic foci demonstrates areas of karyorrhexis, pyknosis, karyolysis in addition to scattered fibrin deposits with collections of the crescentic histiocytes (Figure 2&3). Plasmacytoid dendritic cells are often found clustered at the margins of the necrotic foci [9,11-13]. Scanty plasma cell will be seen if not absent. In addition, the absence of neutrophils and eosinophils is a distinguished characteristic of this syndrome. A mixture of large to small lymphocytes are also present. The numerous presences of large lymphocytes with immunoblastic morphology in a background of karyorrhectic debris and scattered tangible body of macrophages, can often mislead the diagnosis towards high-grade lymphoma [9,11-13]. These necrotic foci may be single or multiple with varying degree of necrosis that differ from case to case [7,9,11-13].
Figure 2:High-power view showing the boundary between an areaof karyorrhexis/pyknosis and an area of karyolysis (H&E, original magnification x100)
Figure 3:Crescentic Histiocytes; background of apoptotic necrosis with abundant karyorrhectic debris (H&E, original magnification x600)
As the disease progress, the histological appearance will also change accordingly. The early proliferative phase is characterized with follicular hyperplasia and paracortical proliferation of T-cells, B-cells, plasmacytoid monocytes, lymphocytes, and histocytes (Figure 4). Numerous apoptosis in the background of this proliferative picture is also a distinguishing feature. As the disease progress to the later necrotizing phase, the histological appearance will progressively change to show necrosis with the absence of neutrophil infiltrate. The presences of numerous crescentic histocytes with or without phagocytosed debris is a distinct feature of the necrotizing phase as well as Kikuchi-Fujimoto disease itself. Finally, with or without necrotic foci, the presence of foamy histocytes indicate the categorization of xanthomatous phase [9,11-13].
Figure 4:Paracortical expansion (H&E, original magnification x100)
Histological differential diagnosis primarily consists of lymphoid malignancy, autoimmune associated lymphadenopathy, and infectious diseases. Systematic exclusion of these entities during the diagnosis of Kikuchi disease is imperative because each of these entities requires disease specific management [9,11-13].
As mentioned previously, the abundance of plasmacytoid dendritic cells and activated T-cell closely mimics the appearance of malignant lymphoma. Necrosis with absence of neutrophil is characteristic of Kikuchi disease. In comparison, necrosis associated with Hodgkin’s lymphoma usually includes numerous neutrophils and eosinophils surrounding large atypical Reed-Sternberg variant cells [12]. These large atypical cells are immunohistochemically positive for CD15, CD30 and PAX-5. The proliferative phase aggregation of immunoblastic proliferation and the necrotic phase plasmacytoid dendritic cells of Kikuchi Fujimoto disease closely resembles the histological presentation of anaplastic large cell lymphoma [9,12]. With the assistance of immunohistology staining, large cell lymphoma can be excluded with the absence of CD30 expression. Similarly, B-cell linage of large cell lymphoma can be excluded with lack of CD20, CD79a, or PAX5 expression. [9,12]. Flow cytometrical analysis may be utilized in difficult and confusing cases.
It is a difficult task to differentiate Kikuchi Fujimoto disease from systemic lupus erythematosus due to near resemblance and occasional identical histological presentation. Useful differentiating features that favors Kikuchi disease includes abundance of CD8 positive cytotoxic T cells around the necrotic areas, scanty plasma cell presence, and absence of neutrophils and eosinophils [7,14]. Differentiating features that favors SLE/autoimmune disease includes the presence of hematoxylin bodies, Azzopardi phenomenon, reactive follicular hyperplasia, scanty cytotoxic t-cells, abundance of plasma cells, and capsular and pericapsular inflammation [7,14]. Due to close histological resemblance and morphology of these two diseases, it is of diagnostic importance, especially in ambiguous situations, to also review patient laboratory data and clinical history [9,11-13]. Infection induced adenopathy will present similar histological features that closely resembles Kikuchi disease. On histological examination, Kikuchi Fujimoto disease can be differentiated from viral lymphadenitis with significant histiocytic infiltrate while lacking in neutrophil aggregation, CD4+ T cell presence, and plasma cell proliferation [12]. Furthermore, due to similar clinical presentation and radiological findings, tuberculosis requires careful differentiation from Kikuchi Fujimoto disease. To differentiate, Kikuchi Fujimoto disease lacks the presence of epithelioid histiocytes, multinucleated giant cells, and granuloma formation found in necrotizing granulomatous lymphadenitis of tuberculosis [11,15].
Kikuchi Fujimoto disease is a self-limiting syndrome. Signs and symptoms usually take four months for full resolution. Recurrence rate is low at around 3-4% [16]. Clinicians should be aware that on rare occasions Kikuchi Fujimoto disease is associated with aggressive life-threatening conditions that include disseminated intravascular coagulation, hemophagocytic syndrome, pulmonary hemorrhage, severe infection, and acute heart failure [16]. Therapy is mainly focused on alleviating symptoms which include relief of fever with antipyretics and reduction of lymph node tenderness with analgesics [9,12,13].
Conclusion
Kikuchi disease, despite its benign lymphadenopathy and self-limited clinical course, requires vigilant examination in order to exclude conditions with overlapping clinical and histological features requiring further aggressive therapy

Read More About this Article: https://biomedgrid.com/fulltext/volume5/a-case-of-kikuchi-fujimoto-disease-histiocytic-necrotizing-lymphadenitis-in-25-years-old-african-american-female.000893.php
For more about: Journals on Biomedical Science :Biomed Grid | Current Issue
#biomedgrid#american journal of biomedical science & research#Health science Journal of Open access#Open access Journals of Biomedical Science
0 notes
Text
Vinblastine Sulfate Market Growth Prospects, Key Vendors and Future Scenario | Global Players - Guangzhou Hanfang Pharmaceutical and Ajinomoto OmniChem, Gedeon Richter Plc, Cipla Inc
Today’s businesses call for the highly focused, comprehensive and detail-oriented information about the market so that they get a clear idea about the market landscape. The Vinblastine Sulfate market research report is generated with a combination of detailed industry insights, and use of latest tools and technology. The study of this market research report covers a market attractiveness analysis, wherein each segment is targeted based on its market size, growth rate & general attractiveness. The Vinblastine Sulfate market research report plays a key role in developing the strategies for sales, advertising, marketing, and promotion.
Get Sample PDF (including COVID19 Impact Analysis) of Market Report @ https://www.databridgemarketresearch.com/request-a-sample/?dbmr=global-vinblastine-sulfate-market
Not to mention, the data included in this Vinblastine Sulfate market document is collected only from the trustworthy sources such as journals, newspapers, company websites and annual reports of the companies on which businesses can rely confidently. Other data models used for the research methodology are vendor positioning grid, market time line analysis, market overview & guide, company positioning grid, company market share analysis, standards of measurement, top to bottom analysis & vendor share analysis. The studies related to competitor analysis in this Vinblastine Sulfate market document keeps competitive landscape clearly into focus with which businesses can decide or advance their own strategies to thrive in the market.
Market Analysis and Insights: Global Vinblastine Sulfate Market
Vinblastine sulfate market is expected to gain market growth in the forecast period of 2020 to 2027. Data Bridge Market Research analyses the market growing at a CAGR of 11.40%% in the above-mentioned forecast period. Data Bridge Market Research report on vinblastine sulfate market provides analysis and insights regarding the various factors expected to be prevalent throughout the forecasted period while providing their impacts on the market’s growth.
The major players covered in the vinblastine sulfate market report are Guangzhou Hanfang Pharmaceutical and Ajinomoto OmniChem, Gedeon Richter Plc, Cipla Inc., Minakem, HISUN USA, inc., Eli Lilly and Company, Vinkem, among other domestic and global players. Market share data is available for global, North America, Europe, Asia-Pacific (APAC), Middle East and Africa (MEA) and South America separately. DBMR analysts understand competitive strengths and provide competitive analysis for each competitor separately.
Get Full TOC, Tables and Figures of Market Report @ https://www.databridgemarketresearch.com/toc/?dbmr=global-vinblastine-sulfate-market
The growing cases of cancer disease are driving the growth of the vinblastine sulfate market. Increasing in-vitro diagnostic tests, curing of histiocytic lymphoma, hodgkin's disease and advanced treatment in cancer patient is driving the growth of the market. The growing innovation and advanced technologies is creating opportunities for the vinblastine sulfate market.
The incidence of side effects such as leucopoenia, blood and lymphatic system disorders and nervous system disorder is acting as a restraint for the market. The usage in elderly patients with cachexia or ulcerated skin acts as a challenge for the market.
This vinblastine sulfate market report provides details of new recent developments, trade regulations, import export analysis, production analysis, value chain optimization, market share, impact of domestic and localised market players, analyses opportunities in terms of emerging revenue pockets, changes in market regulations, strategic market growth analysis, market size, category market growths, application niches and dominance, product approvals, product launches, geographical expansions, technological innovations in the market. To gain more info on vinblastine sulfate market contact Data Bridge Market Research for an Analyst Brief, our team will help you take an informed market decision to achieve market growth.
Global Vinblastine Sulfate Market Scope and Market Size
Vinblastine sulfate market is segmented on the basis of type and application. The growth amongst these segments will help you analyse meagre growth segments in the industries, and provide the users with valuable market overview and market insights to help them in making strategic decisions for identification of core market applications.
Based on type, the vinblastine sulfate market is segmented into type I, type II and type III.
On the basis of application, the vinblastine sulfate market is segmented into I, II and III.
Vinblastine Sulfate Market Country Level Analysis
Vinblastine sulfate market is analysed and market size insights and trends are provided by country, type and application as referenced above.
The countries covered in the vinblastine sulfate market report are the U.S., Canada and Mexico in North America, Germany, France, U.K., Netherlands, Switzerland, Belgium, Russia, Italy, Spain, Turkey, Rest of Europe in Europe, China, Japan, India, South Korea, Singapore, Malaysia, Australia, Thailand, Indonesia, Philippines, Rest of Asia-Pacific (APAC) in the Asia-Pacific (APAC), Saudi Arabia, U.A.E, South Africa, Egypt, Israel, Rest of Middle East and Africa (MEA) as a part of Middle East and Africa (MEA), Brazil, Argentina and Rest of South America as part of South America.
North America dominates the vinblastine sulfate market due to the growing cases of cancer and presence of better facilities in the region, while Asia-Pacific is expected to grow at the highest growth rate in the forecast period of 2020 to 2027 due to growing population advancement in healthcare industries.
The country section of the vinblastine sulfate market report also provides individual market impacting factors and changes in regulation in the market domestically that impacts the current and future trends of the market. Data points such as consumption volumes, production sites and volumes, import export analysis, price trend analysis, cost of raw materials, down-stream and upstream value chain analysis are some of the major pointers used to forecast the market scenario for individual countries. Also, presence and availability of global brands and their challenges faced due to large or scarce competition from local and domestic brands, impact of domestic tariffs and trade routes are considered while providing forecast analysis of the country data.
Healthcare Infrastructure growth Installed base and New Technology Penetration
Vinblastine sulfate market also provides you with detailed market analysis for every country growth in healthcare expenditure for capital equipment, installed base of different kind of products for vinblastine sulfate market, impact of technology using life line curves and changes in healthcare regulatory scenarios and their impact on the vinblastine sulfate market. The data is available for historic period 2010 to 2018.
Competitive Landscape and Vinblastine Sulfate Market Share Analysis
Vinblastine sulfate market competitive landscape provides details by competitor. Details included are company overview, company financials, revenue generated, market potential, investment in research and development, new market initiatives, global presence, production sites and facilities, production capacities, company strengths and weaknesses, product launch, product width and breadth, application dominance. The above data points provided are only related to the companies’ focus related to vinblastine sulfate market.
Customization Available : Global Vinblastine Sulfate Market
Data Bridge Market Research is a leader in advanced formative research. We take pride in servicing our existing and new customers with data and analysis that match and suits their goal. The report can be customised to include price trend analysis of target brands understanding the market for additional countries (ask for the list of countries), clinical trial results data, literature review, refurbished market and product base analysis. Market analysis of target competitors can be analysed from technology-based analysis to market portfolio strategies. We can add as many competitors that you require data about in the format and data style you are looking for. Our team of analysts can also provide you data in crude raw excel files pivot tables (Factbook) or can assist you in creating presentations from the data sets available in the report.
Do You Have Any Query Or Specific Requirement? Ask to Our Industry Expert @ https://www.databridgemarketresearch.com/inquire-before-buying/?dbmr=global-vinblastine-sulfate-market
About Data Bridge Market Research:
Data Bridge Market Research is a versatile market research and consulting firm with over 500 analysts working in different industries. We have catered more than 40% of the fortune 500 companies globally and have a network of more than 5000+ clientele around the globe. Our coverage of industries include Medical Devices, Pharmaceuticals, Biotechnology, Semiconductors, Machinery, Information and Communication Technology, Automobiles and Automotive, Chemical and Material, Packaging, Food and Beverages, Cosmetics, Specialty Chemicals, Fast Moving Consumer Goods, Robotics, among many others.
Data Bridge adepts in creating satisfied clients who reckon upon our services and rely on our hard work with certitude.We are content with our glorious 99.9 % client satisfying rate.
Contact Us :
Data Bridge Market Research
US: +1 888 387 2818
UK: +44 208 089 1725
Hong Kong: +852 8192 7475 Mail: [email protected]
0 notes
Text

Cytotoxic T cells that react to EBV also look like “owl eyes,” apparently. But whenever I hear “owl eyes,” I think of the Reed-Sternberg cells of Hodgkin lymphoma.
From Wikipedia:
Reed–Sternberg cells (also known as lacunar histiocytes for certain types) are distinctive, giant cells found with light microscopy in biopsies from individuals with Hodgkin lymphoma. They are usually derived from B lymphocytes, classically considered crippled germinal center B cells. In the vast majority of cases, the immunoglobulin genes of Reed-Sternberg cells have undergone both V(D)J recombination and somatic hypermutation, establishing an origin from a germinal center or postgerminal center B cell. Despite having the genetic signature of a B cell, the Reed-Sternberg cells of classical Hodgkin lymphoma fail to express most B-cell–specific genes, including the immunoglobulin genes. The cause of this wholesale reprogramming of gene expression has yet to be fully explained. It presumably is the result of widespread epigenetic changes of uncertain etiology, but is partly a consequence of so-called “crippling” mutations acquired during somatic hypermutation. Seen against a sea of B cells, they give the tissue a moth-eaten appearance.[1]
Reed–Sternberg cells are large (30–50 microns) and are either multinucleated or have a bilobed nucleus with prominent eosinophilic inclusion-like nucleoli (thus resembling an "owl's eye" appearance). Reed–Sternberg cells are CD30 and CD15 positive, usually negative for CD20 and CD45. The presence of these cells is necessary in the diagnosis of Hodgkin lymphoma – the absence of Reed–Sternberg cells has very high negative predictive value. The presence of these cells is confirmed mainly by use of biomarkers in immunohistochemistry. They can also be found in reactive lymphadenopathy (such as infectious mononucleosis immunoblasts which are RS like in appearance, carbamazepine associated lymphadenopathy) and very rarely in other types of non-Hodgkin lymphomas. Anaplastic large cell lymphoma may show RS-like cells as well.
2 notes
·
View notes
Text
Neoclomide 50mg 30pcs
Indications Malignant Diseases:
Cyclophosphamide is indicated for the treatment of:
Malignant lymphomas (Stages III and IV of the Ann Arbor staging system), Hodgkin’s disease, lymphocytic lymphoma (nodular or diffuse), mixed-cell type lymphoma, histiocytic lymphoma, Burkitt’s lymphoma
Multiple myeloma
Leukemias: chronic lymphocytic leukemia, chronic granulocytic leukemia (it is usually…
View On WordPress
0 notes
Text
Tổng quan về bệnh ung thư hạch
Ung thư hạch là một loại ung thư máu bắt đầu trong các tế bào chống nhiễm trùng của hệ thống miễn dịch trong cơ thể (hệ thống bạch huyết), được gọi là tế bào lympho.
Khoảng một nửa số bệnh ung thư máu xảy ra mỗi năm là u lympho hoặc ung thư hệ bạch huyết.
Những tế bào này nằm trong các hạch bạch huyết, lá lách, tuyến ức, tủy xương và các bộ phận khác của cơ thể. Khi bạn bị ung thư hạch, tế bào lympho thay đổi và phát triển ngoài tầm kiểm soát.
Có hai loại ung thư hạch chính:
Lymphoma Non-Hodgkin (NHL): Lây lan qua hệ bạch huyết một cách không trật tự. Hầu hết những người bị ung thư hạch đều có loại này.
Lymphoma Hodgkin (HL): Lây lan một cách có trật tự từ một nhóm các hạch bạch huyết khác.
Ung thư hạch có thể xảy ra ở mọi lứa tuổi, nhưng nó là một trong những nguyên nhân gây ung thư phổ biến nhất ở trẻ em và thanh niên từ 15 đến 24 tuổi.
Ung thu hach la gi Lam the nao nhan biet duoc ung thu hach (1)
Ung thư hạch Hodgkin là gì?
Mô bạch huyết có ở nhiều nơi trên cơ thể bạn, vì vậy ung thư hạch Hodgkin có thể bắt đầu bất cứ nơi nào trong hệ thống bạch huyết và phát triển ở nhiều nơi trong cơ thể cùng một lúc. Ung thư hạch Hodgkin thường bắt đầu trong tế bào lympho B.
Mặc dù ung thư hạch Hodgkin có thể bắt đầu ở hầu hết mọi nơi trên cở thể nhưng hầu hết thường bắt đầu ở các hạch bạch huyết ở phần trên của cơ thể. Các mô bạch huyết phổ biến nhất là ở ngực, cổ hoặc dưới cánh tay.
Ung thư hạch Hodgkin thường lây lan qua các mạch bạch huyết gần đó. Nó có thể xâm lấn vào máu và lan sang các bộ phận khác của cơ thể, chẳng hạn như gan, phổi hoặc tủy xương.
Các loại ung thư hạch Hodgkin
Các loại ung thư hạch Hodgkin khác nhau có thể phát triển và lan rộng khác nhau và có thể được điều trị khác nhau.
Ung thư hạch cổ điển Hodgkin
Các tế bào ung thư trong cHL được gọi là tế bào Reed-Sternberg. Những tế bào này thường là một loại tế bào lympho B bất thường. Các hạch bạch huyết mở rộng ở những người mắc bệnh cHL thường có một số lượng nhỏ tế bào Reed-Sternberg với rất nhiều tế bào miễn dịch bình thường xung quanh chúng. Những tế bào miễn dịch khác gây ra hầu hết sưng trong các hạch bạch huyết.
Có 4 kiểu ung thư hạch cổ điển Hodgkin:
Bệnh xơ cứng hạch Hodgkin (NSCHL): Đây là loại bệnh Hodgkin phổ biến nhất nó thường bắt đầu trong các hạch bạch huyết ở cổ hoặc ngực. Bệnh này phát triển phổ biến nhất ở thanh thiếu niên và thanh niên.
Tế bào hỗn hợp Ung thư hạch Hodgkin (MCCHL): Đây là loại bệnh phổ biến thứ hai nó thường bắt đầu ở bất kỳ hạch bạch huyết nào nhưng thường xảy ra ở nửa trên của cơ thể. Nó được thấy chủ yếu ở những người bị nhiễm HIV.
Ung thư hạch Hodgkin giàu tế bào lympho: Bệnh này không phổ biến nó thường xảy ra ở nửa trên của cơ thể và hiếm khi được tìm thấy ở nhiều hơn một vài hạch bạch huyết.
Ung thư hạch Hodgkin bị suy giảm tế bào lympho: Đây là một dạng bệnh hiếm gặp nó được thấy chủ yếu ở người già và những người nhiễm HIV. Nó hung dữ hơn các loại HL khác và có khả năng tiến triển khi lần đầu tiên được tìm thấy. Nó thường gặp nhất ở các hạch bạch huyết ở bụng cũng như ở lá lách, gan và tủy xương.
Hạch lympho Hodgkin chiếm ưu thế
Hạch lympho Hodgkin chiếm ưu thế tế bào lympho (NLPHL) chiếm khoảng 5% trường hợp. Các tế bào ung thư trong NLPHL là các tế bào lớn gọi là tế bào bỏng ngô vì giống như bỏng ngô, là biến thể của tế bào Reed-Sternberg. Nó cũng được gọi là tế bào lymphocytic và histiocytic (L & H).
NLPHL thường bắt đầu trong các hạch bạch huyết ở cổ và dưới cánh tay. Nó có thể xảy ra ở mọi người ở mọi lứa tuổi, và phổ biến ở nam giới hơn nữ giới. Loại HL này có xu hướng phát triển chậm hơn và được đối xử khác với các loại cổ điển.
Ung thư hạch không Hodgkin là gì?
Ung thư hạch không Hodgkin (NHL) là một loại ung thư bắt đầu trong các tế bào bạch cầu gọi là tế bào lympho, là một phần của hệ thống miễn dịch của cơ thể. NHL là một thuật ngữ được sử dụng cho nhiều loại ung thư hạch khác nhau, tất cả đều có chung một số đặc điểm.
NHL ảnh hưởng đến hệ thống bạch huyết của cơ thể. Nó có thể bắt đầu bất cứ nơi nào trong cơ thể, nơi mô bạch huyết được tìm thấy, các mô bạch huyết chính là hạch bạch huyết, lá lách, tủy xương, tuyến ức, đường tiêu hóa, Adenoids & amidan
Nó thường bắt đầu trong các hạch bạch huyết hoặc mô bạch huyết khác, nhưng đôi khi nó có thể ảnh hưởng đến da.
Các loại ung thư hạch không Hodgkin
Điều trị NHL tùy thuộc vào loại nào, vì vậy điều quan trọng là tìm ra loại ung thư hạch chính xác mà bạn có. Loại ung thư hạch phụ thuộc vào loại tế bào lympho bị ảnh hưởng, các tế bào trưởng thành như thế nào khi chúng trở thành ung thư và các yếu tố khác.
Tế bào Lympho
Hệ thống bạch huyết được tạo thành chủ yếu từ các tế bào lympho, một loại tế bào bạch cầu giúp cơ thể chống lại nhiễm trùng. Có 2 loại tế bào lympho chính:
Tế bào lympho B: Tế bào B tạo ra protein gọi là kháng thể giúp bảo vệ cơ thể khỏi vi trùng. Các kháng thể gắn vào vi trùng, đánh dấu để phá hủy chúng bởi các bộ phận của hệ thống miễn dịch.
Tế bào lympho T: Có nhiều loại tế bào T. Một số tế bào T tiêu diệt vi trùng hoặc tế bào bất thường trong cơ thể. Các tế bào T khác giúp tăng cường hoặc làm chậm hoạt động của các tế bào hệ thống miễn dịch khác.
Ung thư hạch không Hodgkin có thể bắt đầu ở một trong hai loại tế bào lympho, nhưng u lympho tế bào B là phổ biến nhất.
U lympho khác
Các loại NHL cũng có thể được nhóm lại dựa trên tốc độ phát triển và lan truyền của chúng:
U lympho không rõ ràng phát triển và lây lan chậm. Một số u lympho không rõ ràng có thể không cần phải điều trị ngay lập tức, nhưng thay vào đó có thể được theo dõi chặt chẽ. Loại ung thư hạch không phổ biến là u lympho nang.
U lympho xâm lấn phát triển và lan rộng nhanh chóng và thường cần phải được điều trị ngay lập tức. Loại ung thư hạch xâm lấn phổ biến nhất là u lympho tế bào B khuếch tán lớn (DLBCL).
Một số loại ung thư hạch, như u lympho tế bào lớp phủ, không phù hợp với một trong hai loại này.
Bất kể chúng phát triển nhanh như thế nào, tất cả các u lympho không Hodgkin đều có thể lan sang các phần khác của hệ thống bạch huyết nếu không được điều trị và chúng cũng có thể lan sang các bộ phận khác của cơ thể, chẳng hạn như gan, não hoặc tủy xương.
Sự khác biệt giữa ung thư hạch Hodgkin và Non-Hodgkin
Sự khác biệt giữa Ung thư hạch Hodgkin và Ung thư hạch không Hodgkin là sự xuất hiệt của các tế bào Reed-Sternberg. Một tế bào Reed-Sternberg là một tế bào có nguồn gốc từ tế bào lympho B và chỉ có trong Ung thư hạch Hodgkin.
Các tế bào Reed-Sternberg được tìm thấy khi khối u được kiểm tra dưới kính hiển vi, được chẩn đoán là ung thư hạch
Nếu không có tế bào Reed-Sternberg trong khối u bạch huyết, chẩn đoán rất có thể là ung thư hạch không Hodgkin.
Trong số tất cả các trường hợp ung thư hạch được chẩn đoán, 90% trong số đó là Ung thư hạch không Hodgkin và 10% Ung thư hạch Hodgkin.
Phân biệt giữa ung thư hạch Hodgkin và ung thư hạch không Hodgkin là rất quan trọng.
Nguyên nhân và Các yếu tố rủi ro
Nguyên nhân
Mô bạch huyết được kết nối khắp cơ thể. Nếu các tế bào ung thư phát triển trong hệ bạch huyết được gọi là tế bào lympho phát triển đột biến gen, chúng có thể dễ dàng lây lan từ vị trí ban đầu của chúng đến các mô và bao gồm cả những bộ phận khác của cở thể.
Nguyên nhân gây ra ung thư hạch là không rõ ràng, nhưng có một số yếu tố nguy cơ cao gây bệnh ung thư hạch.
Các yếu tố rủi ro
Hai loại ung thư hạch khác nhau có các yếu tố nguy cơ khác nhau.
Lymphoma Non-Hodgkin
Các yếu tố nguy cơ của ung thư hạch không Hodgkin bao gồm:
Tuổi tác: Hầu hết các u lympho xảy ra ở những người từ 60 tuổi trở lên, nhưng một số loại có nhiều khả năng ảnh hưởng đến trẻ em và thanh niên.
Giới tính: Một số loại có nhiều khả năng ở phụ nữ, một số khác có nhiều khả năng ở nam giới.
Dân tộc và vị trí địa lý: Ở Mỹ, người Mỹ gốc Phi và người Mỹ gốc Á có nguy cơ mắc ung thư hạch không Hodgkin thấp hơn người Mỹ da trắng, và nó phổ biến hơn ở các quốc gia phát triển.
Hóa chất và phóng xạ: Bức xạ hạt nhân và một số hóa chất được sử dụng trong nông nghiệp có thể gây ung thư hạch không Hodgkin.
Hệ miễn dịch yếu: Một người có hệ miễn dịch yếu có nguy cơ cao hơn. Ví dụ: người nhiễm HIV & AIDS, các loại thuốc được sử dụng sau khi cấy ghép nội tạng.
Bệnh tự miễn: Đây là khi hệ thống miễn dịch tấn công các tế bào của chính cơ thể. Ví dụ: bao gồm viêm khớp dạng thấp và bệnh celiac.
Nhiễm trùng: Một số bệnh nhiễm virus và vi khuẩn làm biến đổi tế bào lympho làm tăng nguy cơ, chẳng hạn như virus Epstein-Barr (EBV), gây ra bệnh sốt tuyến.
Cân nặng và chế độ ăn uống: Béo phì có liên quan đến sự phát triển của ung thư hạch, mặc dù cần nhiều nghiên cứu hơn để xác nhận mối liên kết.
Ung thư hạch Hodgkin
Các yếu tố nguy cơ của bệnh ung thư hạch Hodgkin bao gồm:
Bệnh bạch cầu đơn nhân truyền nhiễm: Nhiễm EBV có thể gây ra bệnh bạch cầu đơn nhân, làm tăng nguy cơ ung thư hạch.
Tuổi tác: Những người trong độ tuổi từ 20 đến 30 và những người trên 55 tuổi có nguy cơ cao hơn.
Giới tính: Nó hơi phổ biến hơn ở nam giới.
Vị trí địa lý: Ung thư hạch Hodgkin phổ biến nhất ở Mỹ, Canada và Bắc Âu. Nó ít phổ biến nhất ở châu Á.
Lịch sử gia đình: Nếu trong gia đình có người được chẩn đoán mắc loại ung thư này, nguy cơ bạn phát triển nó cũng cao hơn.
Nhiễm HIV: Điều này có thể làm suy yếu hệ thống miễn dịch và tăng nguy cơ ung thư hạch.
Dấu hiệu và triệu chứng
Những người mắc ung thư hạch có thể gặp các dấu hiệu và triệu chứng khác như:
Mệt mỏi
Sốt và ớn lạnh
Đổ mồ hôi đêm
Giảm cân
��n mất ngon
Ngứa
Đau cổ hoặc sườn
Bị sưng hạch không đau ở nách, cổ hoặc háng.
Mở rộng của lá lách.
Nếu hạch bạch huyết bị ảnh hưởng nằm trong ngực, nó có thể ảnh hưởng đến hơi thở của người đó; nếu nó ở trong bụng, nó có thể gây khó chịu ở bụng.
Ung thư hạch đôi khi có thể khó chẩn đoán vì những dấu hiệu và triệu chứng này thường nhẹ. Một số người có thể không có dấu hiệu đáng chú ý trong khi những người khác có thể bị sốt nhẹ. Thông thường có các hạch bạch huyết bị sưng, chúng nằm khắp cơ thể thường ở cổ, háng, bụng hoặc nách.
Các giai đoạn của ung thư hạch
Cả NHL và HL có thể được phân thành bốn giai đoạn. Tình trạng ung thư hạch được xác định bởi vị trí của ung thư và mức độ lan rộng của nó hoặc không lan rộng.
Giai đoạn 1: Ung thư nằm trong một hạch bạch huyết hoặc một cơ quan trích dẫn.
Giai đoạn 2: Ung thư nằm trong hai hạch bạch huyết gần nhau và ở cùng một phía của cơ thể hoặc ung thư nằm trong một cơ quan và các hạch bạch huyết gần đó.
Giai đoạn 3: Tại thời điểm này, ung thư nằm trong các hạch bạch huyết ở cả hai bên của cơ thể và trong nhiều hạch bạch huyết.
Giai đoạn 4: Ung thư có thể ở trong một cơ quan và lan ra ngoài các hạch bạch huyết gần đó. Khi NHL tiến triển, nó có thể bắt đầu lan rộng. Các trang web phổ biến nhất cho NHL tiên tiến bao gồm gan, tủy xương và phổi.
Tuy nhiên ung thư hạch giai đoạn 4 tiến triển, nó vẫn có thể điều trị được.
Ung thu hach la gi Lam the nao nhan biet duoc ung thu hach (3)
Điều trị
Bác sĩ sẽ thực hiện sinh thiết hạch để chẩn đoán ung thư hạch. Các xét nghiệm bổ sung sau đó được tiến hành để xác định giai đoạn của ung thư hạch bao gồm:
xét nghiệm máu,
sinh thiết tủy xương
xét nghiệm hình ảnh, chẳng hạn như chụp CT hoặc chụp PET.
Quyết định về điều trị sau đó được xác định bởi bác sĩ của bạn, sẽ xem xét tuổi, sức khỏe chung, giai đoạn và loại ung thư hạch. Ung thư hạch Hodgkin là một trong những loại ung thư có khả năng chữa khỏi cao.
Các lựa chọn điều trị bao gồm:
Hóa trị
Hóa trị và xạ trị nhắm trực tiếp vào ung thư hạch
Các liệu pháp sinh học, như kháng thể, hướng vào các tế bào ung thư hạch
Ghép tế bào gốc
Đối với một số bệnh nhân, tham gia vào một thử nghiệm lâm sàng cung được sử dụng các liệu pháp thử nghiệm.
Nếu bạn được chẩn đoán mắc bệnh ung thư hạch, hãy nói chuyện với bác sĩ về việc tham gia thử nghiệm lâm sàng có phù hợp với bạn hay không.
Phòng ngừa
Bởi vì nguyên nhân của ung thư hạch vẫn chưa được biết, không có cách nào thực sự để ngăn chặn nó. Tuy nhiên, nếu bạn nghĩ rằng bạn có thể có dấu hiệu ung thư hạch, nhận thức được các yếu tố nguy cơ và triệu chứng và nói chuyện với bác sĩ là rất quan trọng để chẩn đoán và điều trị sớm.
Điều đặc biệt quan trọng nếu bạn có tiền sử gia đình mắc bệnh ung thư hạch để tìm ra các triệu chứng và chia sẻ tình trạng y tế của gia đình bạn và bạn với bác sĩ.
>>Thuốc điều trị ung thư hạch: Thuốc Adcetris, Thuốc Opdivo 100mg, Thuốc Opdivo 40mg, Thuốc Leukeran, Thuốc Keytruda, Thuốc Gleostine.
#gallery-0-5 { margin: auto; } #gallery-0-5 .gallery-item { float: left; margin-top: 10px; text-align: center; width: 33%; } #gallery-0-5 img { border: 2px solid #cfcfcf; } #gallery-0-5 .gallery-caption { margin-left: 0; } /* see gallery_shortcode() in wp-includes/media.php */
Thuoc Adcetris 50mg Brentuximab dieu tri ung thu hach Hodgkin – Gia Adcetris (4)
Thuốc Opdivo 40mg10ml Nivolumab điều trị ung thư trúng đích – Giá Thuốc Opdivo
Thuốc Opdivo 100mg/10ml Nivolumab điều trị ung thư trúng đích – Giá Thuốc Opdivo
Thuoc Keytruda Penbrolizumab thuoc dieu tri ung thu tphcm ha noi da nang can tho (4)
Thuoc Leukeran 2mg Chlorambucil dieu tri ung thu mau – gia thuoc Leukeran tphcm hanoi (2)
Thuoc gleostine dieu tri khoi u nao gia thuoc gleostine (2)Thuoc gleostine dieu tri khoi u nao gia thuoc gleostine (2)
>>>Xem thêm: https://nhathuoclanphuong.com/ung-thu-hach-la-gi-lam-the-nao-nhan-biet-duoc-ung-thu-hach/
Ung thư hạch là gì? Làm thế nào nhận biết được ung thư hạch? Tổng quan về bệnh ung thư hạch Ung thư hạch là một loại ung thư máu bắt đầu trong các tế bào chống nhiễm trùng của hệ thống miễn dịch trong cơ thể (hệ thống bạch huyết), được gọi là tế bào lympho.
#bệnh học lympho#lympho bào là gì#lymphoma Hodgkin#lymphoma non Hodgkin#u lympho Hodgkin#u lympho không Hodgkin#ung thư hạch#ung thư hạch bạch huyết
0 notes
Text
Cancers, Vol. 11, Pages 244: Mouse-Derived Isograft (MDI) In Vivo Tumor Models I. Spontaneous sMDI Models: Characterization and Cancer Therapeutic Approaches
Cancers, Vol. 11, Pages 244: Mouse-Derived Isograft (MDI) In Vivo Tumor Models I. Spontaneous sMDI Models: Characterization and Cancer Therapeutic Approaches
Cancers doi: 10.3390/cancers11020244
Authors: Peter Jantscheff Janette Beshay Thomas Lemarchand Cynthia Obodozie Christoph Schächtele Holger Weber
Syngeneic in vivo tumor models are valuable for the development and investigation of immune-modulating anti-cancer drugs. In the present study, we established a novel syngeneic in vivo model type named mouse-derived isografts (MDIs). Spontaneous MDIs (sMDIs) were obtained during a long-term observation period (more than one to two years) of naïve and untreated animals of various mouse strains (C3H/HeJ, CBA/J, DBA/2N, BALB/c, and C57BL/6N). Primary tumors or suspicious tissues were assessed macroscopically and re-transplanted in a PDX-like manner as small tumor pieces into sex-matched syngeneic animals. Nine outgrowing primary tumors were histologically characterized either as adenocarcinomas, histiocytic carcinomas, or lymphomas. Growth of the tumor pieces after re-transplantation displayed model heterogeneity. The adenocarcinoma sMDI model JA-0009 was further characterized by flow cytometry, RNA-sequencing, and efficacy studies. M2 macrophages were found to be the main tumor infiltrating leukocyte population, whereas only a few T cells were observed. JA-0009 showed limited sensitivity when treated with antibodies against inhibitory checkpoint molecules (anti-mPD-1 and anti-mCTLA-4), but high sensitivity to gemcitabine treatment. The generated sMDI are spontaneously occurring tumors of low passage number, propagated as tissue pieces in mice without any tissue culturing, and thus conserving the original tumor characteristics and intratumoral immune cell populations.
http://bit.ly/2IpdJ2Z
0 notes
Text
J chain and myocyte enhancer factor 2B are useful in differentiating classical Hodgkin lymphoma from nodular lymphocyte predominant Hodgkin lymphoma and primary mediastinal large B-cell lymphoma
Although most classical Hodgkin lymphomas (CHL) are easily distinguished from nodular lymphocyte predominant Hodgkin lymphoma (NLPHL) and primary mediastinal large B-cell lymphoma (PMBL), cases with significant CD20 expression cause diagnostic confusion. Although the absence of OCT-2 and BOB.1 are useful in these circumstances, a variable proportion of CHL are positive for these antigens. We investigated the utility of J chain and MEF2B in the diagnosis of CHL, NLPHL, PMBL, T-cell/histiocyte-rich large B-cell lymphoma (TCRLBL), and B-cell lymphoma, unclassifiable, with features intermediate between diffuse large B-cell lymphoma and CHL (BCLU, DLBCL/CHL) compared to OCT-2 and BOB.1. from # All Medicine by Alexandros G. Sfakianakis via alkiviadis.1961 on Inoreader http://ift.tt/2w891y9
from OtoRhinoLaryngology - Alexandros G. Sfakianakis via Alexandros G.Sfakianakis on Inoreader http://ift.tt/2vzd6Id
0 notes