#splenomegaly
Explore tagged Tumblr posts
Visit Tumblr Blog
Explore Tumblr blogs with no restrictions, modern design and the best experience.
Last Seen Tumblr Blogs





Fun Fact
BuzzFeed published a report claiming that Tumblr was utilized as a distribution channel for Russian agents to influence American voting habits during the 2016 presidential election in Feb 2018.
Text
I. Polycythemia Diagnosis:
A. Clinical Assessment:
• The diagnostic process for polycythemia commences with a thorough examination of medical history and physical condition to detect signs indicative of erythrocytosis, assess potential risk factors (e.g., smoking habits, family history of thrombosis), and uncover underlying causes.
• Special attention should be given to symptoms of hyperviscosity syndrome (e.g., headaches, dizziness), skin manifestations (e.g., erythromelalgia), and signs of organ enlargement (e.g., splenomegaly, hepatomegaly).
B. Laboratory Tests:
• Laboratory investigations are crucial for diagnosing polycythemia and understanding its underlying mechanisms. Essential tests include a complete blood count (CBC) with differential, examination of peripheral blood smear, and measurement of serum erythropoietin levels.
• Additional tests, such as JAK2 mutation analysis, bone marrow biopsy, and molecular testing for other mutations associated with myeloproliferative neoplasms (e.g., CALR, MPL), may be necessary to confirm the diagnosis of PV and rule out alternative causes.
C. Imaging Techniques:
• Imaging methods like ultrasound, computed tomography (CT), or magnetic resonance imaging (MRI) may be used to evaluate organ enlargement (e.g., splenomegaly, hepatomegaly) and identify potential underlying factors for secondary polycythemia (e.g., renal tumors).
II. Polycythemia Treatment:
Management of polycythemia aims to alleviate symptoms, lower the risk of thrombotic complications, and prevent disease progression. Treatment approaches may vary depending on the underlying cause and severity, often involving a combination of strategies:
A. Phlebotomy (Venesection):
• Phlebotomy is the primary treatment for PV, involving the removal of excess blood to achieve target hematocrit levels (<45% in men, <42% in women).
• Regular phlebotomy sessions usually start at diagnosis and are adjusted based on individual response and disease activity.
B. Cytoreductive Therapy:
• Drugs like hydroxyurea, interferon-alpha, and ruxolitinib may be used in PV patients who do not respond to or cannot tolerate phlebotomy.
• These drugs work by suppressing abnormal hematopoietic proliferation and reducing the risk of blood clotting, with hydroxyurea being the most commonly used and studied cytoreductive drug in PV.
C. Antiplatelet Therapy:
• Aspirin and other antiplatelet drugs are often prescribed to PV patients with a history of blood clots or other high-risk factors to lower the risk of arterial thrombosis.
• Aspirin is usually started at low doses (e.g., 81 mg daily) and may be combined with cytoreductive therapy for better thromboprophylaxis.
D. Treating Underlying Conditions:
• Management of secondary polycythemia focuses on addressing the root cause to relieve hypoxia-induced erythropoiesis and prevent disease progression.
• Interventions may include oxygen therapy for patients with chronic respiratory problems, correction of hemoglobin disorders or other genetic issues, and surgical removal of erythropoietin-secreting tumors.
E. Lifestyle Changes:
• Lifestyle adjustments such as quitting smoking, maintaining a healthy weight, regular physical activity, and proper hydration are vital for improving clinical outcomes and reducing cardiovascular risks in polycythemia patients.
Doctors suggest undergoing regular health checkups for the early diagnosis and treatment of polycythemia. You can choose to undergo a regular full body health checkup at Jaslok Hospital Mumbai, which is one of India's best hospitals for the early detection and management of blood disorders.
#polycythemia#headache#dizziness#CBC#biopsy#splenomegaly#hepatomegaly#phlebotomy#cytoreductive therapy#hematopoietic proliferation#aspirin#antiplatelet therapy#hypoxia#respiratory problems#blood disorders#full body health checkup#regular health checkups
0 notes
Text
Exploring Effective Treatments for Spleen Diseases

Discover comprehensive approaches and innovative therapies for managing various spleen diseases. From conventional medical interventions to emerging treatments, learn about options to improve patient outcomes and enhance quality of life. Explore the evolving landscape of spleen disease treatment strategies and their potential impact on patient care and well-being.
#spleen Diseases Treatment#spleen Diseases Symptoms#splenomegaly causes#enlarged spleen diseases#spleen enlargement#enlargement of spleen treatment#spleen diseases and symptoms#enlarged spleen splenomegaly#causes enlarged spleen
0 notes
Text
1 note
·
View note
Text


**Health update**
So I never wanted to make another post like this but here I am.. I was hoping to just get better and never bring it up again.
To get those of you up to speed. I was diagnosed with CKD back in late 2021. I’ve been battling that for a while now. I’m actually doing really well in that regard. During a routine MRI in Nov 2023 for my kidneys we found what looked to be a hepatic steatosis and splenomegaly. It was later determined and I was diagnosed with hepatic tumors and splenomegaly. I also had a fairly large gallstone and what appeared to be a tumor/cyst on my gallbladder. We decided it was just better to remove my GB and the tumors all at the same time. So in January of this year I had laparoscopic cholecystectomy/tumor removal. That’s what the scars on my stomach are from. The tumors were tested and were benign. That was a huge relief. But it left my liver fairly damaged. I wasn’t to concerned about because your liver can and will heal itself. I recovered well and felt great.
Fast forward 9 months. I wasn’t feeling very well. So I made an appointment with my PCP and she wanted me to do a bloodwork panel. This is normal. We actually do blood once a month but really only check kidneys, thyroid, and hormones. So I did the panel and when the test results came in my Dr called me immediately. She ordered another AST/hepatic function panel for my liver, kidneys and pancreas.
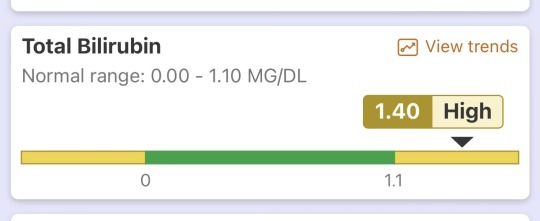
After those test results came in she had me come to her office. I was met with her and a Hepatologist. They broke the news to me that unfortunately my liver not doing well according to the bloodwork. My pancreas/kidneys are actually pretty ok rn which is cool. But my total bilirubin is 1.4 mg/dl. At 1.5 mg/dl total bilirubin is where we really start looking at the possibility of Cirrhosis. If you don’t know what that is. Google it. Now I’m not saying I have this rn but it seems to be looking that way 🫤
They were telling me they could see my bilirubin trending up over the course of the past few months of bloodwork. Also previously being diagnosed with Splenomegaly which is a tell tale sign of Cirrhosis apparently. They’re pretty sure I have it or will have it. They want to run more tests and see how bad or good it is and we’ll go from there. It seems like they have a pretty good plan. Also for those of you that are going to say get a second opinion.. I got a second and a third. They said the same thing go figure..
So again I find myself going in and out of the hospital again. I don’t wish this life upon anyone. It’s not fun. Being chronically/terminally ill is no fucking joke. If it’s not one thing it’s another. It’s tiring and I’m soo tired. Honestly I can’t wait until it’s all over.. forever.
Anyway I figured I’d give you a bit of an update on my health and such. A lot of you ask in my DMs. It’s hard to answer everyone so a post like this gets it out there. But yea if you made it this far thank you and I love you 🫶🏻
#im sick#still sick#always will be sick#if it’s not one thing it’s another#health#chronic illness#terminally ill#please dont feel sorry for me#I don’t want that#trans#transgender#trans pride#transisbeautiful#mtf#transgirl#girlslikeus#mtf hrt#maletofemale#transformation#trans woman#trans women#trans women are beautiful#transexual#actually trans#this is what trans looks like#trans people#trans positivity#mtf positivity#actually mtf#mtf pride
167 notes
·
View notes
Text
Why the Spleen Sucks
The spleen is a really shittily placed organ, making it prone to injury. This injury is usually severe and can lead to death if not properly managed. We're going to look at the function of the spleen, what happens when it is damaged, and how to write about.
Where is the spleen? It's in the upper left quadrant of the abdominal cavity, nestled right against the ribs (typically 9-11) at the midaxillary line. It's behind the stomach and is considered intraperitoneal. The main thing is that the spleen is very vulnerable. It is literally right up against the ribs without much protecting it. It's shaped like a little bean and is purple in humans. It is fed by the splenic artery, which comes off of the celiac trunk (which sticks off of the abdominal aorta).
What does the spleen do? Its main job is to filter out old and malformed red blood cells. It also holds immune cells. Certain diseases can cause the spleen to enlarge, including cirrhosis of the liver (it's connected to the hepatic portal system), sickle cell anemia (RBCs are stuck in it), and autoimmune disorders. The spleen also holds about 250 mL of RBCs in reserve in case you need them.
What happens when it is injured? The spleen can be ruptured and lacerated kinda easily. Blunt trauma to the ribs can cause it to rupture, and this is seen in contact sports and car accidents mostly. Because of those giant gaps between the ribs, it's also prone to injury from knife attacks. Gunshot wounds are another common cause, as well as broken ribs penetrating it (broken ribs are very sharp, like way sharper than you imagine). Rupture is more likely when someone has splenomegaly.
When the spleen is damaged, you're going to get a lot of intraperitoneal hemorrhaging. The spleen filters a lot of blood and has blood in it, so there's going to be a lot of blood in the abdomen (obviously). This will lead to distention, guarding (abs are tense), and hypovolemia. The left upper quadrant will be painful, and there can also be referred pain to the left shoulder (Kehr's sign).
If the patient has a small laceration, the symptoms aren't always as dramatic. Sometimes they'll just have low hemoglobin (which is on RBCs), maybe some thrombocytopenia (lots of platelets in the blood).
How do you fix this? If the injury is small and the patient is hemodynamically stable, they can usually be given a blood transfusion and the spleen can heal itself. Sometimes surgery is also performed to clamp a vessel or repair the outer layer of the spleen.
If the injury is major, then surgery will be performed. If the patient is less critical, they may go in and try to fix the problem. If it can't be fixed, they may do a splenectomy (remove the spleen). In a critical patient, they might forgo the nice pretty incision on the left side, and instead just split the patient down the middle. In these situations (in my experience), there isn't a lot of time to waste. One thing that we aren't going to waste time on is anesthesia, for example. This is with a lot of very critical surgeries, at least from what I have seen. Like the surgeon will start cutting as they are working on knocking out the patient, but usually they are in so much pain that they don't even register it.
If you remove the spleen, the patient is more at risk for infections, but with modern medicine and vaccinations, it's not as much of a big deal as it used to be. The patient will probably be fine.
Writing tips: (new section idea, hope you guys like it, lol) As with any injury, you have to make sure that you are giving them an acceptable mechanism of injury. With the spleen, this is either blunt trauma or penetration/laceration. Getting tackled, getting stabbed, getting shot, all great MOIs.
Second thing, present the appropriate signs and symptoms. A sign would be like bruising, hypotension, tachycardia, etc. A symptom would be LUQ pain, Kehr's sign, etc.
Next, figure out what you're going to do and where you're going to do it. In the field, there probably isn't much you can do. The most would probably be a laparotomy and clamping the splenic artery, but I mean, when I was an EMT, we were not doing this. There's a lot of stuff you can theoretically do, but never gets done. But I mean you can write it. If the patient makes it to the hospital, I think it would be more fun to do emergency surgery and just split them right down the middle. There's going to be a lot of blood in the greater omentum, very high stakes and exciting.
Anyways, hope you guys liked this, please let me know if I got anything wrong. I wrote this off of my personal experience and a few good textbooks, but there can always been mistakes in things.
#medicine#med student#medical school#biology#med school#med studyblr#whump writing#anatomy#spleen#hospital whump#surgery#emergency medicine#medical writing#writing reference#injury
75 notes
·
View notes
Text
having the hEDS trifecta + lupus is very frustrating because it's just like "okay where is this symptom coming from?" and no doctor can agree so it doesn't get treated
I have professionally diagnosed MCAS
I'd like to share the symptoms of MCAS can cause:
Constitutional : Fatigue, subjective hyperthermia and/or hypothermia, sweats, change in appetite, weight gain/loss, chemical/physical sensitivities, poor healing
Dermatologic : Urticaria, itch, flushing, hemangiomas with itch/pain, various rashes, telangiectasias, striae, skin tags, folliculitis, ulcers, eczema, angioedema, alopecia, onychodystrophy
Ophthalmologic : Irritated, “dry” eyes, difficulty focusing, blepharospasm
Otologic : Tinnitus, hearing loss, coryza, rhinitis, nasal congestion, epistaxis
Oropharyngeal: Pain, burning, leukoplakia, ulcers, angioedema, dysgeusia, dental and/or periodontal inflammation/decay
Lymphatic: Lymphadenopathy, rare splenomegaly
Pulmonary: Dry cough, dyspnea (difficulty taking a deep breath), wheezing, obstructive sleep apnea
Cardiovascular: Presyncope, hypertension, blood pressure lability, palpitations, edema, chest pain, allergic angina (Kounis syndrome)
Gastrointestinal: Dyspepsia, gastroesophageal reflux, abdominal pain, nausea, vomiting, diarrhea and/or constipation, gastroparesis, angioedema, dysphagia (usually proximal), bloating (post-prandial or spontaneous), malabsorption
Genitourinary: Menorrhagia, pelvic pain, endometriosis, vulvodynia, vaginitis, dysmenorrhea, miscarriages, infertility, dysuria
Musculoskeletal: Myalgias, migratory bone/joint pain, osteopenia/osteoporosis
Neurologic : Headache, migraine, sensory neuropathies, dysautonomia, episodic weakness, seizure disorders, non-epileptic seizures, cognitive dysfunction, insomnia, hypersomnolence, restless leg syndrome
Psychiatric: Depression, anger/irritability, mood lability, anxiety, panic, obsession–compulsion, attention deficit/hyperactivity
Hematologic: Easy bruising, polycythemia, anemia
Immunologic: Hypersensitivity reactions, increased risk for malignancy and autoimmunity, impaired healing, increased susceptibility to infection
source
"well that's just everything" ya. that's why I can't figure out where my symptoms are coming from. lupus is also like this. autonomic neuropathy is also like this. my doctors run in circles pointing at the other doctors to solve the problem but none of them want to.
#physical disability#physically disabled#chronic illness#chronically ill#mast cell activation syndrome#mast cell disease#mast cell activation disorder#hypermobile ehlers danlos syndrome#hEDS#systemic lupus erythematosus#dysautonomia#pots syndrome#postural orthostatic tachycardia syndrome#autonomic neuropathy
27 notes
·
View notes
Text
Q Fever
Aka, Query fever. What a weird name for a disease. Imagine telling people that's what you got.

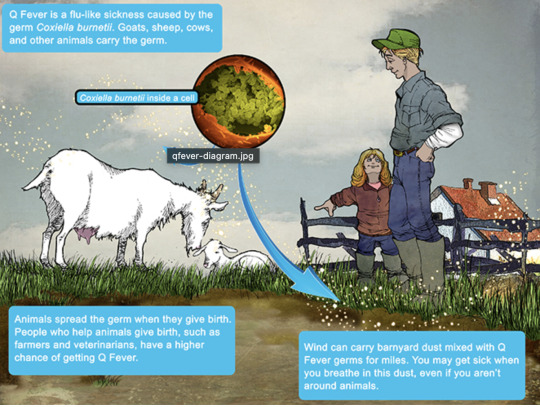
in the 30s-40s, an Australian pathologist in QLD/Brisbane, came across an outbreak of the same or similar illness among abbatoir or slaughterhouse workers.
At the time, he called the disease "Q" fever or query as a temporary name until the pathogen could be identified. Unfortunately it stuck.
decades later, now nobel prize winner and virologist, MacFarlane Burnett isolated and identified the microbe responsible. I think this discovery contributed to his prize. i forget already.
Microbe responsible: Coxiella burnetti. Named for Burnett and HR Cox, the American bacteriologist who found the genus Coxiella where C burnetti falls under.
Initially they felt it was related to Rickettsia, responsible for Rocky Mountain Spotted Fever, but as science progressed, this was disproven.
Now for a Case Report
A 55 yo Italian man with a history of aortic valve replacement was diagnosed with pyrexia of unknown origin twice. Further signs included myalgias/splenomegaly/night sweats. The 2nd time he was admitted for PUO he deteriorated rather dramatically and was put on meropenem and teicoplanin.
A host of organisms was tested for on serological testing based on the man's travel and epidemiological history, all negative. Even a rheumatological panel was done, also less revealing. He also had a history of MGUS (a haem disoder), which is kind of a red herring here.
Cultures were negative, no vegetations were seen on a TTE - so they did consider IE. Which is an important differential for PUO.
Eventually a PET-CT was done (often favoured when investigations do not yield much for a sick patient with fevers), finally revealing a focus of infectious on his ascending aorta, where he'd also had previous surgery done. And in a round about way, they also further identified Coxiella Burnetti. He was treated doxycycline and hydroxychloroquine. As it's so rare in Italy, it wasn't really considered even though he mentioned rural travel.
Bottomline: Q Fever is an important consideration in the work up for culture negative IE. Further to this, always consider IE in the differentials for PUO particularly if they're at increased risk for IE (prosthetic valves, damaged valves, select congenital heart issues, previous IE). IE can present with night sweats, fevers, weight loss and splenomegaly. It can be insidious and chronic in nature. other risk factors can be more suggestive as we'll get into below.
Causative organism
Coxiella burnetti, it's a zoonoses - i.e. transmissible from animals. Special powers: very tough/hardy, can survive extreme environments (high temps and UV light etc.) over prolonged periods and is resistant to many common disinfectants/surface cleaners.
It's an intracellular pathogen and gram negative coccobacilli (PINK!)
name coccobaccili reminds me of cocopuffs.

it's mainly associated with farm animals, which the CDC so wholesomely displays on its website on Q fever (wtf).

goats, sheep, cattle typically (but many other animals, even birds, dogs and horses can be reservoirs)
in particular bodily fluids - amniotic fluid, placenta, faeces/urine, milk etc.
you can get it through unpasteurized milk and through inhaling it if it lands on dust in the area
ever visit a farm or petting zoo lately? OMG WASH YOU HANDS.

That said, it's typically inhaled in inorganic dust. You inhale it, it goes to the lungs, and then the bloodstream.
Increased risk for Coxiella burnetti (What to take on history of exposures and when to strongly consider it)
live on a farm or near one
exposure to a farm
work as a vet on a farm
farm worker, dairy workers, researchers on these animals/facilities
slaughterhouse/abbatoir
Also from CDC:
Clinical presentation
Most won't get sick after exposure and remain asymptomatic, a very small minority does. even though it is highly infectious.
incubation time is 2-3 weeks (consider this time in your history of exposure, did they work on the farm 2-3 weeks ago as opposed to yesterday).
Nonspecific acute infectious symptoms:
nonspecific systemic fevers/malaise/arthralgias/myalgias--> key is high fevers though and can be associated with headache and photophobia.
non specific GI - N/V/diarrhoea
respiratory ones - SOB or cough, consider it as atypical cause of community acquired pneumonia.
rare: hepatitis and jaundice (granulomatous) or encephalitis with neurological complications such as demyelinating disease or CN palsies, also haemolytic anaemia and HLH (yikes)

really it's the history of exposure that will lead you down the garden path to Q fever.
Chronic Q fever is perhaps worse, and can present as culture negative IE/PUO. Months/years later, as B symptoms as above above + LOW/LOA, night sweats. More likely to occur if you are predisposed for IE as above, have a weakened immune system for any reason, including pregnancy.
Chronic Q fever has a mortality of 10% if left untreated. About <5% of those with acute Q fever develop this if left untreated. Speculation is that it's more of an autoimmune process or abnormal immunological response to the bacteria.
To be honest, most who walk in the door with community acquired pneumonia get treated empirically for atypicals anyway, (standard course of doxycycline), so we hardly really ponder the question of Q fever in every patient. But if they present chronically and did not have atypical cover at the onset of acute symptoms, then it's something important to consider.
Other important conditions - can cause complications in pregnant women and 20% will get post Q fever syndrome. like chronic fatigue.
investigations
Serology! nice and easy. Look for IgG antibodies in the chronic presentation. Or PCR. Down side to serology - can take 2-3 days for the body to make said antibodies to the bacteria for detection. PCR can be done on any fluids/tissue sent.

Cultures useless, hence it fall under the umbrella of culture negative (hard to grow outside a host cell, it is an obligate intracellular pathogen).
Other hints on bloods (as serology/PCR takes time to return) - elevated or low platelet's, transaminitis with normal bili, opacities in CXR with hilar lymphadenopathy, CSF will show raised protein levels if done when encephalitis is suspected.
imaging can also support the diagnosis.. as illustrated by the case report.
Treatment
Acute disease - as standard for atypical bugs, doxycycline 100 mg BD for 14 days. Alternatives - TMP SMX or Clarithromycin.
Chronic Q fever or IE:
native valves: doxycycline and hydroxychloroquine (200 TDS) for 18 months
prosthetic: same but 24 months
why hydroxy: enhances the action of doxycycline (increases the pH of the phagolysosome)
Follow-up: look for 4 fold decrease in IGG
Sources:
CDC
Stat Pearls
Wiki as linked above
#australian history#medblr#medblrs#infectious disease#infectious diseases#q fever#coxiella#coxiella burnetti
74 notes
·
View notes
Text
Reference archived on our website
Abstract
Objective COVID-19 induces the development of autoimmune diseases, including SLE, which are characterised by inflammation, autoantibodies and thrombosis. However, the effects of COVID-19 on SLE remain unclear.
Methods We investigated the effects of COVID-19 on SLE development and progression in three animal models. Plasmids encoding SARS-CoV-2 spike protein and ACE2 receptor were injected into R848-induced BALB/C lupus mice, R848-induced IL-1 receptor antagonist knockout (KO) lupus mice and MRL/lpr mice. Serum levels of albumin and autoantibodies, lymphocyte phenotypes and tissue histology were evaluated.
Results In R848-induced BALB/C lupus mice, the SARS-CoV-2 spike protein increased autoantibody and albumin levels compared with vehicle and mock treatments. These mice also exhibited splenomegaly, which was further exacerbated by the spike protein. Flow cytometric analysis revealed elevated T helper 1 cell counts, and histological analysis indicated increased levels of the fibrosis marker protein α-smooth muscle actin. In KO mice, the spike protein induced splenomegaly, severe kidney damage and pronounced lung fibrosis. In the MRL/lpr group, spike protein increased the serum levels of autoantibodies, albumin and the thrombosis marker chemokine (C-X-C motif) ligand 4.
Conclusion COVID-19 accelerated the development and progression of lupus by inducing autoantibody production, fibrosis and thrombosis.
#long covid#lupus#covid#covidー19#mask up#pandemic#covid 19#wear a mask#public health#coronavirus#sars cov 2#still coviding#wear a respirator#covid isn't over#covid conscious#covid is airborne#covid pandemic#covid19
16 notes
·
View notes
Text
I have a pt who has alpha thalassemia minor in her medical problem list. She had asked for blood tests because she felt anemic. Her Hgb/hct are normal. Iron panel showed low iron saturation and elevated UIBC.
Unsaturated iron binding capacity (UIBC) is a blood test that measures the amount of iron that can still bind to transferrin in the blood. It's part of a panel of tests that evaluate iron levels.
What's it used for?
UIBC is used to assess iron deficiency or overload.
It's often ordered along with a serum iron test and a total iron-binding capacity (TIBC) test.
UIBC can help diagnose conditions like anemia, hemochromatosis, and iron toxicity.
What do UIBC results mean?
Elevated UIBC: May indicate iron deficiency or malabsorption.
Decreased UIBC: May indicate iron overload, hemochromatosis, or other conditions.
When might a UIBC test be ordered?
If a complete blood count (CBC) shows low hemoglobin and hematocrit
If there are signs of anemia, like chronic fatigue, dizziness, or weakness
If there are signs of iron overload, like joint pain, weight loss, or low sex drive
If there's a family history of hemochromatosis
Basically, she has weird hemoglobin so she can't use iron properly. I don't think I need to actually do anything unless she becomes anemic. I just referred her to heme/onc.
Looking on UpToDate, basically alpha thalassemia minor would now be called non-transfusion dependent. Beta thalassemia is transfusion dependent. They need blood transfusions. This is what UTD says about thalassemia management:
Anemia – The main treatments for anemia are transfusions and luspatercept (algorithm 1). Transfusions reduce symptoms and morbidities of anemia and ineffective erythropoiesis. Ineffective erythropoiesis can impair growth and development and cause skeletal abnormalities, splenomegaly, and iron overload. (See 'Management of anemia' above.)
•Transfusion-dependent – For individuals with TDT (previously called thalassemia major phenotype), we suggest chronic transfusion (Grade 2C). (See "Diagnosis of thalassemia (adults and children)", section on 'Overview of subtypes and disease severity' and 'Regular transfusions' above.)
We generally suggest a pretransfusion hemoglobin of 9.5 to 10.5 g/dL rather than a higher or lower nadir (Grade 2C). Clinical experience and observational studies demonstrate this hemoglobin nadir is associated with greater survival, provided iron stores are controlled, and reduced extramedullary hematopoiesis. Different hemoglobin nadirs may be appropriate in individuals with certain comorbidities or without clinically significant ineffective erythropoiesis. (See 'Decision to initiate regular transfusions' above and 'Typical chronic transfusion regimen' above.)
Luspatercept can reduce transfusion requirements in some adults with transfusion-dependent beta thalassemia (TDT). Some individuals may choose to take luspatercept, and others may continue with regular transfusions while awaiting more information on long-term outcomes. (See 'Luspatercept for transfusion-dependent thalassemia' above.)
•Non-transfusion-dependent – For non-transfusion-dependent thalassemia (NTDT, previously called thalassemia intermedia phenotype), management is individualized. Most patients need only periodic transfusions for symptomatic relief or during periods of stress (rapid growth, infection-associated bone marrow suppression, surgery, pregnancy). Some patients may become require regular transfusions (see 'Decision to initiate regular transfusions' above). The role of erythropoiesis-modifying agents is unknown.
•Thalassemia minor – Chronic transfusions are not required when anemia is very mild or absent.
•Alpha thalassemia major – Management is discussed separately. (See "Alpha thalassemia major: Prenatal and postnatal management".)
For individuals with hemolytic anemia, we also suggest folic acid (Grade 2C). Folic acid may reasonably be omitted if especially burdensome and folate levels are adequate. (See 'Dietary restrictions and supplements' above and "Clinical manifestations and diagnosis of vitamin B12 and folate deficiency", section on 'Diagnostic evaluation'.)
●Excess iron stores – Patients receiving transfusions require regular assessment and treatment of excess iron stores. (See 'Assessment of iron stores and initiation of chelation therapy' above and "Iron chelation: Choice of agent, dosing, and adverse effects".)
●Splenectomy – Splenectomy is an option for severe anemia, hypersplenism, or other splenic complications; but we avoid splenectomy when possible. The benefit may be transient, and risks (life-threatening infection; thromboembolism, especially in patients receiving luspatercept) are increased. When pursued, splenectomy is generally deferred until ≥4 years. Pre-splenectomy vaccines and infection and thromboembolism are required. (See 'Role of splenectomy' above.)
●Transplantation – Allogeneic hematopoietic stem cell transplantation is potentially curative and may be appropriate for severe TDT (algorithm 1). The decision to pursue transplantation is complex and should be made in consultation with a thalassemia specialist and experienced high-volume transplant center. (See 'Decision to pursue allogeneic HSCT' above.)
●Monitoring – Individuals with TDT are typically seen by a specialist at least two to four times yearly for monitoring and management of iron stores and other disease manifestations (table 3). (See 'Monitoring and management of disease complications' above.)
●Curative and investigational therapies – Several curative therapies (allogeneic hematopoietic stem cell transplantation, gene therapy) and medical therapies are under investigation, and two gene based therapies have been approved for TDT but not for alpha thalassemia.
•Considerations including when to offer curative therapies, how to choose among the different options, and how to optimize health and iron status prior to these therapies, are discussed separately. (See "Hematopoietic stem cell transplantation and other curative therapies for transfusion-dependent thalassemia".)
•Investigational medical therapies are discussed above. (See 'Investigational approaches' above.)
●Reproductive counseling – Genetic testing and reproductive counseling is routine for all individuals with thalassemia. (See 'Reproductive testing and genetic counseling' above and "Hemoglobinopathy: Screening and counseling in the reproductive setting and fetal diagnosis".)
●Prognosis – Survival continues to improve into the fourth, fifth, and sixth decades of life. Cardiovascular complications are the major cause of death in TDT. (See 'Prognosis' above.)
3 notes
·
View notes
Text
G6PD Deficiency
Glucose-6-phosphate-dehydrogenase, commonly called G6PD is an enzyme that is essential for the normal functioning of red blood cells and plays an important role in the oxidization process to maintain the physiology of our body.
G6PD deficiency is genetic and it is characterized by the X- linked metabolic disorder. The deficiency is followed by a premature breakdown of red blood cells, that is the rate of haematogenesis is less than hemolysis. Due to the rapid destruction of RBCs, hemoglobin found in RBCs fails to carry the oxygen from the lungs to different organs of the body resulting in hemolytic Anemia.
Cause of G6PD Deficiency
G6PD deficiency is caused by hereditary factors only, no external stimuli contribute to its deficiency.
Symptoms G6PD Deficiency
In the majority of cases, a person does not show any symptoms and remains a carrier of G6PD deficiency for a prolonged time unless he is exposed to certain food or drugs like fava beans, sulpha drugs, or any noxious agent. Generally, symptoms appear when there is the rapid destruction of RBCs, in such situation patient shows the symptoms of hemolytic anemia like the appearance of pale skin, shortness of breath, abdominal pain with backache, splenomegaly, dark urine, tachycardia, headache with dizziness.
Infants with G6PD deficiency show signs of jaundice 1-4 days after birth due to rapid destruction of RBCs, though mild jaundice is a physiological phenomenon noted in newborns, but the prolongation of complaint may suggest G6PD deficiency.
Nutritional Management :
Though G6PD deficiency is genetically caused, there are some supportive nutritional measures to combat the ill effects of hemolytic anemia, these are:
Avoid eating fava beans which can lead to hemolytic anemia.
Avoid using high doses of ascorbic acid supplementation as it leads to hemolysis.
avoid consuming soya products.
Any edible items containing menthol should be avoided.
Increase consumption of food rich in antioxidants like tomatoes, apples, oranges, etc.
Promote the consumption of whole grains like oats, millet, and barley to get an adequate amount of carbohydrates.
A diet rich in vitamins and folic acid should be consumed rather than taking additional supplements.
Excessive amounts of vitamin K supplements have been proven to cause hemolytic anemia.
Scope of homeopathy :
Homeopathy has a significant effect on genetic diseases like G6PD deficiency. The holistic approach of homeopathy helps in the reversibility of disease conditions and the restoration of health back to its normal state. Homeopathic medicines do not have a direct action on a particular part or organ but it acts on the altered immunity and allow the body to heal itself without causing any side effects.
The single remedy thus selected after taking the totality of symptoms into consideration, arrest the progress of premature hemolysis caused by G6PD deficiency and boost the hemoglobin to carry the oxygen from the lungs to another part of the body, thereby promoting the process of oxidization and prevent the complications caused by hemolysis.
There are many homeopathic medicines that act well in hemolytic anemia caused by G6PD deficiency. Some of them are as follows :
Ferrum met: This remedy is indicated when the person has a great weakness. There is paleness of whole skin with pseudo flushes on slight excitement. Headache is pulsating in character with vertigo and ringing in the ears. Shortness of breath with tachycardia is predominantly present.
Aletris farinosa: This remedy acts well when there is extreme weakness and tired feeling throughout the day with a pale face. The patient feels complete loss of energy and powerlessness. Frequent episodes of giddiness and faintness are present.
Ferrum Phos: This remedy can be easily used for people of all age groups. The patient presents with complaints of paleness, and generalized weakness with nocturnal perspiration. There is tachycardia with an increased pulse rate.
Natrum mur: This remedy is useful when hemolytic anemia is associated with weight loss. The patient appears lean, thin, and malnourished with a loss of vital energy. Hemolysis cause a bursting type of headache with nausea and vomiting.
Calcarea Phos: This remedy is indicated when there is anemia with a slow recovery. There is a tendency to develop bone-related issues. A patient has brittle bones, weakness and difficulty in concentration.
Conclusion
In conclusion, Homeo Care Clinic offers a holistic approach to treating G6PD deficiency. The remedies mentioned above can treat the underlying causes of the condition and offer relief from the discomfort. However, it is important to consult a qualified homeopathic practitioner for the correct dosage and duration of treatment. Homeo Care Clinic provides comprehensive care for various ailments, including G6PD deficiency, and offers customized treatment plans based on individual requirements.
To schedule an appointment or learn more about our services, please visit our website or give us a call. Our friendly staff will be happy to assist you.
Follow us on Facebook, Twitter, and Instagram for valuable insights into the world of homeopathy and holistic health.
4 notes
·
View notes
Text
What are 3 diseases that affect the spleen?

Splenomegaly, characterized by an enlarged spleen, can stem from diverse causes like infections (such as mononucleosis or malaria), liver ailments (like cirrhosis), blood disorders (like sickle cell anaemia), or specific cancers. Symptoms might not always manifest, but when severe, they can include abdominal pain, early satiety, or increased susceptibility to bleeding.
#enlarged spleen splenomegaly#enlargement of spleen treatment#spleen diseases#spleen cancer#spleen pain treatment#splenomegaly Doctors in Ahmedabad
0 notes
Text
Familial lipoprotein lipase (LPL) deficiency, also known as familial chylomicronemia syndrome (FCS), is a rare genetic disorder characterized by a deficiency or dysfunction of the lipoprotein lipase enzyme. This enzyme is responsible for breaking down triglycerides in the bloodstream. In individuals with familial LPL deficiency, the impaired enzyme function leads to the accumulation of chylomicrons (a type of lipoprotein) and very high levels of triglycerides in the blood. The symptoms of familial LPL deficiency may include:
Pancreatitis: Recurrent episodes of pancreatitis, which is inflammation of the pancreas, are a hallmark feature of familial LPL deficiency. Pancreatitis can cause severe abdominal pain, nausea, vomiting, and potentially life-threatening complications.
Lipemia Retinalis: Lipemia retinalis is a condition characterized by creamy or milky-white appearance of the retinal blood vessels. It occurs due to the presence of excess chylomicrons in the blood, which can affect the appearance of the retinal blood vessels during eye examination.
Xanthomas: Xanthomas are fatty deposits that can develop under the skin or around tendons due to the accumulation of lipids. In familial LPL deficiency, xanthomas may appear as yellowish nodules or plaques on the skin, typically on the elbows, knees, buttocks, or feet.
Abdominal Pain: Abdominal pain and discomfort, which may be severe, can occur due to pancreatitis or other digestive disturbances associated with high triglyceride levels.
Hepatomegaly: Hepatomegaly refers to an enlarged liver. In familial LPL deficiency, hepatomegaly can occur due to the accumulation of triglycerides in liver cells.
Splenomegaly: Splenomegaly refers to an enlarged spleen. In some cases of familial LPL deficiency, an enlarged spleen may be present due to the increased workload of the organ in clearing chylomicrons from the bloodstream.
Recurrent Lipid-Related Symptoms: Individuals with familial LPL deficiency may experience recurrent episodes of abdominal pain, nausea, vomiting, and other symptoms related to high triglyceride levels.
2 notes
·
View notes
Text
oh i forgot to worry about the splenomegaly. glad i remembered
2 notes
·
View notes
Text
highlights from the abdominal CT i had last week

[Spleen: 1.1 cm hypodense lesion in the medial spleen is likely a benign hemangioma. No splenomegaly.]
neat, i have a little friend in my spleen

[Reproductive organs: Uterus is normal in size.]
brother i fuckin hope not lol, i paid good money for that to no longer be the case
2 notes
·
View notes
Text

Dalak Büyümesi, Splenomegali: 5 Nedeni ve Tedavisi Dalak büyümesi ya da tıbbi literatürdeki adıyla splenomegali, vücudun sol üst kısmında yer alan ve bağışıklık sisteminde önemli rolleri bulunan dalak organının normal boyutlarının üzerine çıkmasıyla ortaya çıkan bir sağlık durumudur. Read the full article
0 notes
Text
Leishmaniasis
Case Reports, like we're on a episode of house

23M in Kenya, presenting with months of LOW, persistent fevers, and abdo fullness, found to have massive splenomegaly.
examination: massive splenomegaly (10 cm below costophrenic margin, and will definitely cross midline) and hepatomegaly
pancytopaenic on bloods, plt's down to 40s
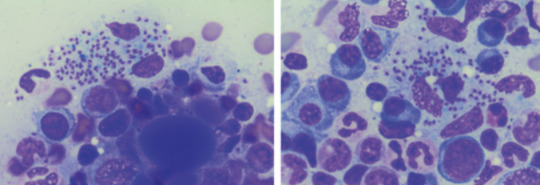
diagnosis confirmed on BMAT (parasite seen)
normal HIV, liver and kidney function
Bodies seen on the BMAT below are part of the lifecycle of the parasite that is intracellular, hence you can see the macrophages/neutrophils loaded with them, even bursting
What is it:
think of it when you get a patient with pancytopaenia and hepatosplenomegaly, who either traveled to or is in/from a tropical/subtropic region (where sand flies are)
cause - protozoa parasite Leishmania, transmitted by infected sandflies
Epidemio (when to consider it)
tropics, subtropics (South America, Asia, AFrica), Southern Europe
Microbiology/Transmission
parasite, replicates intracellularly (Leishmania donovani)
transmitted in sand flies (can be unnoticeable and usually bite in dawn or dusk - evenings or night), can also be transmitted via needles/blood
more common in rural areas
I've simplified this, but is more extensively covered in StatPearls and Wiki (there's different species of Leish and sandflies that transmit it)
once bitten, the protozoa are phagocystosed by skin macrophages, which then becomes full of the "bodies" (part of the lifecycle). Eventually these burst to release more of the bodies that infect more macrophages
they eventually are spread via blood to liver/spleen/BM and LNs
Random history:
ancient, records of disease date back to Egyptian mummies from 3000 BC --> positive DNA amplication for Leishmania and on papyrus from 1500 BC

multiple physicians from different times have described the disease, but it's named for 2 who described the parasite's intracellular ovoid body stage in smears from infected patients in India: Lt General William Boog Leishman and Captain Charles Donovan (Ronald Ross named the bodies after the 2 --> "Leishman Donovan bodies"
significant disease in Allied troops in Sicily in WWII, called "jericho buttons" (image on wiki from a WWI trooper serving in the middle east)

Leishman: Scottish pathologist and British Army medical officer, later it's director general in the 20s, did extensive research into the parasite named for him by Sir Ronald Ross. He mistook the parasite he observed for trypanosomes (cause of Chagas in South America and African sleeping sickness in Africa)
Donovan: Irish parasitologist, medical officer in India, observed an epidemic across India just after the rebellion of 1857, discovered the "bodies" in spleen tissue as the causative agent for what the locals called "kala azar" (severe visceral leishmaniasis - see below)
Donovan also discovered the "bodies" of Klebsiella granulomatis, hence these too are named after him (cause of ulcerative granulomas)
It became scandalous as both wanted credit for the "discovery" of this newly identified organism. So Sir Ronald Ross named it for both of them.
Sir Ron, by the way, won a Nobel in Medicine for discovering that malaria is transmitted via mossies (this was also a source of scandal, he was meant to share it with another physician who he accused of fraud - and they never received the award)
finally, it was actually a Russian physician who identified it first, but well, he published in a little known Russian journal which was promptly forgotten.
Clinical features
cutaneous type vs visceral organ type (spleen, liver, bones)
From wiki
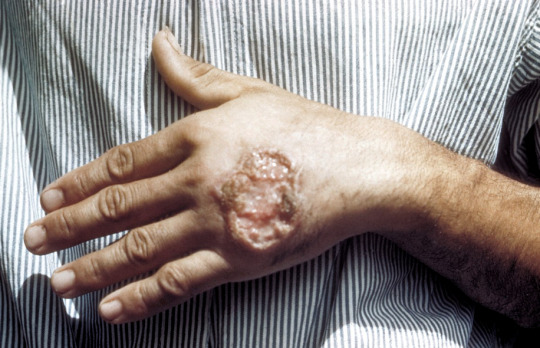
can be asymptomatic
cutnaeous: can be there for years and resemble leprosy, causes an open chronic wound (most common), incubation 2-4 weeks on average (nodules at site of inoculation that eventually form ulcers), can heal spontaneously in 2-5 yrs
in diffuse cutaneous cases, can affect face, ears, extensor surfaces
can be muscosal = eg nasal symptoms/epistaxis, severe: perforated septum, this occurs in 1/3 after resolution of cutaenous symptoms (can be severe/lifte threatning, as it can affect vocal cords and cartilage, but oddly not bone)
visceral (incubation periods of up to years until immuncompromise): fever, weight loss, hepatosplenomegaly (spleen more than liver), pancytoaepnia, high total protein and low albumin with hypergammaglobulinaemia
this has seasonal peaks related to sandfly habits and humidity
interestingly it is an infective cause of massive splenomegaly, such that it crosses the midline
Extreme - but noticeable hepatosplenomgealy/abdo fullness, from medscape

can be atypical in HIV co infected patients, LAD in seom regions like Africa
Kala azar = black fever in some severe cases (fatal due to secondary mycobacterial infection or bleeding), refers to damage fto spleen, liver and anaemia
invstigations:
serology not great (minimal humoral response to the parasite), so often requires histopath (tissue sample) for which BMAT is safest in visceral organ involvement
visualisation of amastigotes (or Leishman-Donovan bodies), as intracellular --> can be seen in macrophages (small round bodies) post Giemsa staining
PCR of DNA also possible (as done in the Egyptian mummies)
Image source:

Treatment

liposomal amphotericin B (holy shit strong stuff) in visceral, PO: miltefosine (caution in pregnancy), all have significant ADRs, or paromycin. however, mortality of 10% if visceral left untreated
mixed results with azoles
in HIV co infection - start the HAARTs! can improve survival, mortality is 30% in HIV patients
cutaneous: stibolgluconate (have never heard of these drugs) and megluaine antimoniate, but limited disease often spotnaeously gets cleared by the innate system
prevention:
use DEET insect repellant at dawn and dusk
loose fitting clothing that covers all skin
no vaccine (were attempts at vaccinating dogs, which decreased rates)
sandflies are smaller than mossies, so requires small netting

Differentials for hepatosplenomegaly

Sources:
WHO guidelines
CDC guidlelines
Wiki - Haven't covered pathophysio, but wiki does extensively
StatPearls
DermNet - great resource for all things derm, that my derm colleagues pointed out to me
9 notes
·
View notes