#liaison ionique
Explore tagged Tumblr posts
Visit Tumblr Blog
Explore Tumblr blogs with no restrictions, modern design and the best experience.
Last Seen Tumblr Blogs





Fun Fact
Tumblr Inc. has $15.1M in annual revenue.
Text
Complexes et ligands : une autre approche
Un composé complexe est constitué de ligands entourant un atome métallique ou un ion métallique central. Dans un post précédent, nous avons présenté la liaison chimique reliant ligand et corps central (appelée liaison de coordination) comme résultant de l'occupation d'une orbitale moléculaire (OM) par un doublet électronique apporté par le ligand. Ce doublet est dit non liant car non engagé dans une autre liaison chimique. Dans cette approche, de nombreux ligands sont censés être des anions : H-, Cl-, CN-, NO2-... La présence d'une charge électrique est dans ce cas indispensable pour justifier la présence du doublet non liant. L'atome d'hydrogène par exemple ne possède qu'un électron et il faut supposer la présence d'anions hydrures H- qui en possèdent deux pour justifier l'existence de complexes comme l'hydrure de fer tétracarbonyle Fe(H)2(CO)4. C'est la raison pour laquelle on qualifie cette approche de ionique, même si de nombreux ligands sont moléculaires (comme le ligand H2O qui possède deux doublets non liants sur son atome d'oxygène).
Approche covalente
De nombreux chimistes lui préfèrent une autre approche, dite covalente. Elle est moins intuitive, mais certainement plus rigoureuse. Elle présente l'avantage de ne pas nécessiter l'intervention d'anions. Le recours à des ligands sous forme d'anions n'a rien d'extravagante lorsqu'il s'agit d’ions halogénures ou cyanures. Ils sont fréquents dans la nature. C'est moins courant pour l'ion hydrure et, en tout état de cause, il n'existe pas d'hydrure de fer FeH2 hormis dans des conditions de température et de pression tout à fait exceptionnelles. Et que dire des complexes organométalliques comme l'hexaméthyl tungstène W(CH3)6 ? Le recours à un ion méthyl CH3- est totalement : le méthane CH4 n’est pas un acide ! Idem pour l’ion cyclopentadiène C5H5-, inconnu au bataillon, nécessaire pour expliquer la structure du ferrocène Fe(C5H5)2 dans l’approche ionique.
L’approche covalente fait l’économie de l’hypothèse ionique en supposant l’existence de deux types de ligands : les ligands L, qui possèdent bel et bien un ou plusieurs doublet non liants, et les ligands X qui sont des radicaux. Un radical est une espèce chimique (atome ou molécule) qui possède un ou plusieurs électrons non appariés sur sa couche électronique externe. On note cet électron non apparié par un point. L’atome d’hydrogène .H est un radical, tout comme l’atome de chlore .Cl (trois doublets et un électron non apparié dans bande de valence) ou la molécule de cyanure .CN. Dans la molécule de cyanure, l’atome de carbone engage trois de ses électrons de valence dans la liaison avec l’azote, il lui reste donc un électron non apparié. Même chose pour le radical méthyl .CH3 puisque trois électrons de la bande de valence du carbone sont engagés dans une liaison covalente avec un atome d’hydrogène. Dans le cas de l’oxygène (1s2, 2s2, 2p4), ce n’est pas un, mais deux électrons de la bande de valence qui sont non appariés (la sous-couche 2p est constituée d’un doublet et deux électrons non appariés). On le note :O. Même chose pour le soufre :S.
Reprenons le cas de l’hexacyanoferrate [Fe(CN)6]4-. Nous avons analysé les liaisons comme intervenant entre un cation fer (II) et des anions cyanure CN-. Dans l’approche covalente, le corps central est un atome de fer non ionisé auquel sont reliés des radicaux cyanure .CN. Dans ce modèle, c’est le complexe hexacyanoferrate considéré comme un corps chimique qui porte la charge -4. Il n’est donc plus question de doublet apporté par le ligand. Dans cette approche, l’atome de fer et les radicaux contribuent à part égale à la liaison de coordination qui les relie. Le fer par un électron de valence, le radical par son électron non apparié.
Equivalence des deux approches
Comme nous l’avons souligné plus haut, l’approche covalente suppose l’existence de deux types de ligands. Les ligands L fournissent le doublet occupant l’OM (comme le ligand H2O ou le ligand CO). Les ligands radicalaires X n’apportent qu’un seul électron à l’OM, leur électron non apparié, l’autre étant apporté par l’atome métallique central. Ceci ne change strictement rien au raisonnement qui conduit à déterminer la géométrie et l’énergie des orbitales moléculaires. Nous allons montrer que le décompte des électrons conduit également au même résultat, tout comme le calcul du degré d’oxydation.
Prenons le cas du dichloro tétraaquo chrome (III) [CrCl2(H2O)4]+. Dans l’approche ionique, on part d’un cation chrome (III) Cr3+, de deux anions chlorure Cl- et de quatre molécules H2O. La charge du complexe se déduit de la charge de ses composants (on utilise le terme fragments) : +3-2=1. Dans l’approche covalente, le corps central est un atome de chrome, il y a deux ligands X qui sont des radicaux .Cl, quatre ligands L qui sont des molécules H2O et une charge électrique nulle.

Dans la première approche, on connait le degré d’oxydation du métal et on en déduit la charge du complexe. Dans la seconde, on part de la charge du complexe pour en déduire le degré d’oxydation du métal. Pour cela, on fait l’hypothèse que tous les ligands, L ou X, sont titulaires du doublet qui assure la liaison (ils sont plus électronégatifs que le métal : ils attirent l’orbitale moléculaire vers eux). Soit x le nombre de ligands X et q la charge du complexe, le degré d’oxydation du métal est égal à x+q. Dans l’exemple du dichloro tétraaquo fer (II), on retrouve bien +3.
Nota : on remarquera que le dichloro tétraaquo chrome (III) ne respecte pas la règle des 18 électrons.
On peut faire le même type de comparaison pour l’ion hexacyanoferrate (formule [Fe(CN)6]4-).

Le degré d’oxydation du fer est par ailleurs égal à 6-4=2. La formule générale du décompte électronique est :
N = m + 2l + x – q
m étant le nombre d’électrons dans la bande de valence du métal, l le nombre de ligands L, x le nombre de ligands X et q la charge du complexe.
Liaisons pi et hapticité
Dans le post précédent, nous avons basé notre présentation essentiellement sur des liaisons sigma entre ligand et corps central (recouvrement d’orbitales orientées dans l’axe passant entre les atomes). Si ce type de liaison est courant en chimie, ce n’est pas le seul. Dans la molécule de monoxyde de carbone CO, le carbone et l’oxygène entretiennent une double liaison. L’une est une liaison sigma, l’autre une liaison pi. Une liaison pi établit un pont entre deux orbitales perpendiculaires à l’axe entre les deux atomes (à condition qu’elles soient dans le même plan). De ce fait, le doublet d’électrons à l’origine de la liaison occupe une position latérale par rapport à la molécule. Ce type de liaison est omniprésente en chimie organique, que ce soit dans les hydrocarbures insaturés ou dans les hydrocarbures aromatiques. C’est le cas par exemple dans la molécule d’éthylène C2H4 (formule semi-développée H2C=CH2), ou dans celle du benzène C6H6. Dans la molécule de benzène, les 6 électrons pi occupent une orbitale délocalisée dont la forme d’ onde est située latéralement par rapport au plan du cycle.
L’orbitale associée à cette liaison pi peut former une orbitale moléculaire liante avec un atome métallique situé lui aussi latéralement par rapport à la molécule. L’éthylène peut donc jouer le rôle de ligand dans un composé complexe bien qu’il ne dispose pas, à proprement parler, de doublet non liant. Le sel de Zeise, ou trichloroéthène platinate (II) de potassium, de formule K[PtCl3(C2H4)]·H2O, en est un exemple. C’est un sel de potassium hydraté dont l’anion est le trichloroéthène platine (II) [PtCl3(C2H4)]-. C’est un complexe plan qui a la forme d’un carré dont les sommets sont occupés par trois ligands chloro (liaison sigma) et un ligand éthène (liaison sigma-pi). On repère ce type de liaison en faisant précéder le nom du ligand par la lettre grecque éta avec en exposant le nombre d’atomes du ligand concernés par la liaison de coordination. Dans le cas du sel de Zeise, ce sera donc éta2 : [PtCl3(éta2-C2H4)]-.

Le benzène se prête quant à lui à des liaisons éta6, comme dans l’ion [Ru(C6H6)(H2O)3]2+. C’est l’orbitale délocalisée du benzène tout entière qui se coordonne avec l’atome de Ruthénium qui se trouve sur un axe perpendiculaire au plan du cycle. On donne le nom d’hapticité au nombre d’atomes du ligand qui se coordonne avec l’atome central au travers de la liaison pi. L’hapticité du ligand éthylène est de 2, celle du benzène de 6.
Le dichloro(cycloocta-1,5-diène) platine(II) PtCl2C8H12 est un autre exemple de ce type de complexe. Le cycloocta-1,5-diène C8H12 est un cyclo-alcène comportant deux doubles liaisons reliant respectivement les carbones 1 et 2 et 5 et 6. L’atome de platine central entretient une liaison de coordination éta2 avec chacune de ces liaisons éthène et la molécule de cyclooctadiène forme un pont entre elles (voir plus bas le paragraphe sur les chélates).
Dans le ferrocène Fe(C5H5)2 (dicyclopentadiényle de fer) l’atome de fer est pris en sandwich entre deux ligands cyclopentadiényle C5H5. Le cyclopentadiényle peut être décrit comme un cycle à cinq carbone comportant deux doubles liaisons (1-2 et 4-5), l’atome de carbone 3 possédant un électron non apparié. Les doublets des liaisons pi et l’électron non apparié (5 électrons en tout) ne sont pas dans le plan du cycle. Ils forment une orbitale pi délocalisée située latéralement qui se coordonne avec l’atome de fer. On peut donc dire que le cyclopentadiényle [C5H5] est un ligand éta5 puisque 5 atomes sont concernés par la liaison de coordination avec l’atome de fer central.
Nota : dans ce cas, il est clair que l’approche covalente est plus adaptée que l’approche ionique. Le cyclopentadiényle est un ligand L2X.

On remarquera que le ferrocène se conforme à la règle des 18 électrons. Le fer compte 8 électrons dans sa bande de valence et chaque ligand cyclopentadiényle en apporte 5 autres, ce qui fait bien 18 électrons. Le ruthénium Ru et l’osmium Os forment le même type de complexe avec des cycles cyclopentadiényle.
Ligands pontants
L’approche covalente s’avère également plus convaincante que l’approche ionique pour expliquer l’existence de ligands pontants. Comme nous l’avons signalé plus haut, un ligand L peut disposer de deux ou plusieurs doublets non liants sur une même atome. C’est le cas par exemple de l’atome d’oxygène dans la molécule H2O ou dans le radical hydroxyle .OH. On dit des ligands H2O et .OH que ce sont des ligands L2. Nous avons également indiqué qu’un ligand X pouvait lui aussi disposer de plusieurs électrons non appariés dans sa couche de valence. C’est le cas de l’atome d’oxygène :O qui est un ligand X2. Le chlore quant à lui dispose d’un électron non apparié mais également de plusieurs doublets non liants : c’est un ligand L2X (le chlore a 7 électrons sur sa bande de valence mais l’un des doublets ne participe pas aux liaisons de coordination pour des raisons de symétrie).
De tels ligands peuvent former une liaison de coordination avec deux (ou plus) atomes métalliques simultanément, ce qui assure une liaison entre eux. Un tel ligand est un ligand pontant. Les ligands pontants établissent un pont entre deux complexes. Ce type de liaison est repérée par la lettre grecque mu. Le ligand hydroxo joue un tel rôle dans la molécule {(Fe(H2O)4)2(mu-OH)2}4+. Elle est constituée de deux édifices octaédriques qui partage l’arête (virtuelle) qui relie les deux sommets OH. Les quatre autres sommets de ces octaèdres sont occupés par des ligands H2O. Le nom de ce nom est tetraaquo fer(3) di-mu-hydroxo tetraaquo fer(III).

Dans le complexe Nb2Cl10, deux atomes de chlore sont pontants et huit sont en position terminale (formule semi-développée (NbCl4)2(mu-Cl)2). On retrouve la même configuration géométrique combinant deux octaèdres. Dans le complexe (RuCl(C6H6))2(mu-Cl)2 chaque atome de ruthénium entretient une liaison éta6 avec une molécule de benzène, une liaison de coordination simple avec un atome de chlore terminal et une liaison pontante avec l’autre atome de ruthénium par le biais de deux autres atomes de chlore.
Nota : les liaisons pontantes au travers d’atomes de chlore vont toujours par deux. De la sorte, un atome de chlore se comporte comme un ligand L avec l’un des atomes métalliques et comme un ligand X avec l’autre (et réciproquement).
Ce type de liaison peut conduire à la constitution d’édifices polyatomiques assez élaborés. Le dodécaméthyl tétrachloro tétraplatine Pt4Cl4(CH3)12 forme un cube dont les huit sommets sont occupés par les quatre atomes de platine et les quatre atomes de chlore. Chaque face du cube est constituée par deux atomes de platine et deux atomes de chlore an position trans (non adjacents). De la sorte, chaque atome de platine est relié à trois atomes de chlore (deux liaisons L et une liaison X). Chaque atome de platine est également relié à trois ligands méthyles. Récapitulons :
chaque atome de platine se coordonne avec 6 ligands, à savoir trois ligands méthyles (ligands X), deux ligands chlore donneurs d’un doublet (ligand L), un ligand chlore donneur d’un électron non apparié (ligand X),
chaque atome de chlore se comporte comme un ligand L2X et fait le pont avec trois atomes de platine.

Le décompte électronique pour chaque atome de platine est le suivant :
m + 2xl + x - q = 10 + 2x2 + (1+3) + 0 = 18
(La structure électronique de la bande de valence du platine est 6s2, 5d8. Le degré d’oxydation du platine est +4.)
Dans le sel noir du Roussin K[Fe4S3(NO)7] on est en présence d’une configuration géométrique originale. Les 4 atomes de fer et les trois ligands X2 :S occupent les sommets d’un cube dont le huitième sommet est vacant. Chaque ligand :S est relié à deux atomes de fer. Une autre particularité de ce complexe est que trois des atomes de fer sont dans la configuration Fe(NO)2S2 et le quatrième (celui qui est diamétralement opposé au sommet vacant) est dans la configuration Fe(NO)S3.

Chélate et denticité
Nous avons vu qu’un même atome au sein d’un ligand pouvait être porteur de plusieurs doublets liants ou de plusieurs électrons non appariés. Un même ligand peut également être porteur de plusieurs atomes susceptibles de se lier à un métal par une liaison de coordination. Le nombre de ces atomes est appelé denticité du ligand. Un ligand peut être bidenté, tridenté, quadridenté... voire même hexadenté. L’exemple le plus connu de ligand bidenté est l’éthylènediammine H2N-CH2-CH2-NH2 (formule condensée C2H8N2). Il peut se coordonner par le biais de ses deux fonctions amine -NH2. Dans le dichloro(éthylènediamine) palladium (II) PdCl2(C2H8N2) l’éthylènediamine se coordonne avec l’atome de palladium par ses deux extrémités amine comme si c’étaient deux ligands L. Un composé complexe au sein duquel l’atome métallique central est relié à un même ligand par deux liaisons de coordination est un chélate.

Le chlorure de dichlorobis(éthylènediammine) cobalt(III) est un sel composé d’un anion chlorure et d’un cation [CoCl2(C2H8N2)2]+. Le cation dichlorobis(éthylènediammine) cobalt(III) a une structure octaédrique : il se coordonne avec deux ligands chlore et deux ligands éthylènediammine par ses deux extrémités amine (ce qui fait 2x2 liaisons de coordination). Il respecte la règle des 18 électrons :
N = 9 + 4x2 + 2 – 1 = 18
L’éthylènediamine tétraacétique C10H16N2O8 (EDTA) est hexadenté : les deux fonctions amine peuvent servir de ligand ainsi que les quatre ions carboxylate (chaque groupe amine est lui-même porteur de deux groupes carboxyliques (goupes -CH2-C(=O)OH). L’EDTA a un pouvoir chelatant fort. On l’utilise comme antidote pour traiter les intoxications par les métaux lourds.
Pour en savoir plus :
post sur la classification périodique des éléments
post sur les liaisons chimiques
post sur les orbitales moléculaires
post sur la valence
post sur la géométrie des molécules
post sur les complexes et les ligands : l’approche ionique
post sur les complexes et les ligands : exemples
post sur l’azote
post sur les acides et les bases
index
#complexe#coordination#chimie#liaison covalente#liaison ionique#chelate#chelation#ligand#denticité#ferrocène#ligand pontant#orbitale moléculaire
0 notes
Text
biochimie, inhibiteurs
L'activité d'une enzyme peut être modulée par d’autres molécules :
un inhibiteur est une molécule qui diminue l'activité d'une enzyme,tandis qu'un
activateur l'accélère ; de nombreux médicaments mais aussi des poisons sont des inhibiteurs enzymatiques.
Un inhibiteur est une molécule, généralement de petite taille, qui lorsqu’elle se lie à une enzyme, en diminue l’activité catalytique.
Elle peut
⚙️empêcher la fixation du substrat en se liant à sa place dans le site actif, ou bien encore
⚙️provoquer une déformation plus ou moins étendue de la structure tridimensionnelle de l'enzyme ne permettant plus d’assurer la catalyse de la réaction.
L’inhibition peut être réversible ou bien irréversible.
Les inhibiteurs dits réversibles se lient aux enzymes par des liaisons non covalentes, de faible énergie, comme des liaisons hydrogène, des liaisons ioniques ou encore des interactions hydrophobes.
L’ensemble de ces liaisons permet d’établir une association plus ou moins forte et plus ou moins spécifique entre l’enzyme et l’inhibiteur.
(tips pour exam) :
Déformation structure 3D enzyme
Réversible liaison h ionique
Permettent liaison enzyme donc modif 3D
Compétitif - ressemblance structurale
Affinité apparente ⬇️vitesse Ma⬆️
Incompéte
Affinité apparente ⬆️vitesse Max⬇️
0 notes
Photo

IRAN GALBANUM OIL Nouvelles: Huile essentielle d'eucalyptus Eucalyptol (1,8 cinéole) de l'huile essentielle d'eucalyptus, un inhibiteur potentiel de l'infection par le virus Corona COVID 19 par les études d'amarrage moléculaire FR: COVID-19, un membre de la famille des virus corona, étend ses tentacules à travers le monde en raison du manque de médicaments à l'heure actuelle. La toux, la fièvre et les problèmes respiratoires sont associés à son infection et provoquent plus de 15% de mortalité dans le monde. Elle est causée par un virus à ARN simple brin positif de la famille des coronavirus enveloppés. Cependant, la principale protéinase virale (Mpro / 3CLpro) a récemment été considérée comme une cible appropriée pour la conception de médicaments contre l'infection par le SRAS en raison de son rôle vital dans le traitement des polyprotéines nécessaires à la reproduction des coronavirus.Objectifs: La présente étude in silico a été conçue pour évaluer la effet de l'eucalyptol (1,8 cinéole), un composant d'huile essentielle de l'huile d'eucalyptus, sur Mpro par étude d'amarrage.Méthodes: dans la présente étude, des études d'amarrage moléculaire ont été menées en utilisant des outils de dock 1-click et swiss dock. Le mode d'interaction des protéines a été calculé par Protein Interactions Calculator.Résultats: Les paramètres calculés tels que RMSD, l'énergie de liaison et la similitude du site de liaison ont indiqué une liaison efficace de l'eucalyptol à la protéinase COVID-19. La prédiction de sites actifs a en outre validé le rôle des résidus de sites actifs dans la liaison des ligands. Les résultats PIC ont indiqué que les complexes Mpro / eucalyptol forment des interactions hydrophobes, des interactions de liaison hydrogène et des interactions ioniques fortes.Conclusions: Par conséquent, l'eucalyptol peut représenter un potentiel de traitement potentiel pour agir en tant qu'inhibiteur de Mpro COVID-19. Cependant, des recherches supplémentaires sont nécessaires pour étudier leur utilisation médicinale potentielle. #huileessentielle #Galbanum #asafoetida #herbals #plants #medicin #medicinal #Meditation #Eucalyptus #relax #Irangalbanumoil #COVID19 https://www.instagram.com/p/B-4oJn7hB-_/?igshid=172tznreyrxf8
#huileessentielle#galbanum#asafoetida#herbals#plants#medicin#medicinal#meditation#eucalyptus#relax#irangalbanumoil#covid19
0 notes
Text
Les toxines
Les toxines sont des substances présentes dans les aliments, l’eau, l’air et le sol.



La résonance induite par une exposition au rayonnement infrarouge long d’une longueur d’onde de l’ordre de 10 micromètres, va mettre les molécules d’eau contenant les toxines en vibration . Cette vibration réduit les liaisons ioniques des atomes qui tiennent ensemble les molécules d’eau. Comme la position des…
View On WordPress
0 notes
Link
Le taux de CO2 atmosphérique ne cessant d’augmenter, les scientifiques cherchent en permanence de nouveaux moyens d’utiliser le dioxyde de carbone en excès afin, entre autre, de régler les différentes questions énergétiques qui se posent actuellement à l’échelle globale. Récemment, une équipe de chercheurs a mis au point un système de photosynthèse artificielle permettant de produire des carburants liquides à partir du C02 présent dans l’atmosphère.
Des chimistes de l’Université de l’Illinois ont réussi à produire des carburants en utilisant de l’eau, du dioxyde de carbone et de la lumière visible, par photosynthèse artificielle. En convertissant le dioxyde de carbone en molécules plus complexes telles que le propane, cette technologie verte ouvre une nouvelle voie vers l’utilisation du CO2 en excès pour stocker l’énergie solaire — sous la forme de liaisons chimiques — utilisable quand l’ensoleillement est faible et que la demande est grande.
Les plantes utilisent la lumière du Soleil pour produire des réactions chimiques entre l’eau et le CO2 afin de créer et de stocker de l’énergie solaire sous forme de glucose dense en énergie. Dans cette nouvelle étude publiée dans la revue Nature Communications, les chercheurs ont mis au point un procédé artificiel utilisant la même gamme de lumière du spectre de la lumière visible utilisé par les plantes lors de la photosynthèse naturelle pour convertir le CO2 et l’eau en carburant, associé à des nanoparticules d’or riches en électrons servant de catalyseur.
« L’objectif ici est de produire des hydrocarbures complexes et liquéfiables à partir de CO2 en excès et d’autres ressources durables telles que la lumière solaire » déclare Prashant Jain, chimiste et auteur de l’étude. « Les carburants liquides sont idéaux car ils sont plus faciles, plus sûrs et plus économiques à transporter que le gaz et, comme ils sont fabriqués à partir de molécules à longue chaîne, ils contiennent plus de liaisons, ce qui signifie qu’ils renferment plus d’énergie ».
Du carburant liquide produit par photosynthèse artificielle
Dans leur laboratoire, les chercheurs utilisent des catalyseurs métalliques pour absorber la lumière et transférer les électrons et les protons nécessaires aux réactions chimiques entre le CO2 et l’eau, remplissant ainsi le rôle du pigment chlorophylle dans la photosynthèse naturelle.
Les nanoparticules d’or fonctionnent particulièrement bien comme catalyseur, explique Jain, car leurs surfaces interagissent favorablement avec les molécules de CO2, absorbent efficacement la lumière et ne se dégradent pas.
Sous la lumière solaire et assistées par un liquide ionique, les nanoparticules d’or (jaune) transfèrent des électrons pour convertir les molécules de CO2 (sphères rouges/grises au centre) en molécules d’hydrocarbures plus complexes. Crédits : Sungju Yu
L’énergie stockée dans les liaisons du combustible hydrocarboné est libérée de plusieurs manières. Cependant, la méthode conventionnelle simple de combustion finit par produire plus de CO2, ce qui va à l’encontre de la notion de récupération et de stockage de l’énergie solaire.
Sur le même sujet : Des structures d’ADN capables de reproduire artificiellement la photosynthèse
« Il existe d’autres utilisations potentielles, non conventionnelles, des hydrocarbures créés à partir de ce processus. Ils pourraient être utilisés pour alimenter des piles à combustible afin de produire du courant et une tension électriques. Il existe des laboratoires dans le monde entier qui tentent de déterminer comment la conversion d’hydrocarbure en électricité peut être conduite efficacement » explique Jain.
Aussi intéressant que le développement de ce combustible CO2-liquide puisse être pour la technologie de l’énergie verte, les chercheurs reconnaissent que le processus de photosynthèse artificielle de Jain est loin d’être aussi efficace que dans les plantes.
« Nous devons apprendre à ajuster le catalyseur pour augmenter l’efficacité des réactions chimiques. Ensuite, nous pourrons entamer le travail difficile consistant à déterminer comment intensifier le processus. Et, comme toute technologie énergétique non conventionnelle, de nombreuses questions de faisabilité économique se poseront également » conclut Jain.
Source: Trust My Science
0 notes
Text
De la confiture de fruits rouges fraises framboises bleuets cassis et l’engouement de la pectine des fruits certaines personnes ajoutent un gélifiant tel que…
youtube
La gelée de framboises est une confiture où n’est pas conservée la pulpe des fruits le jus de framboises ingrédients pour 8 barres 1 banane.
Dans le boutehors le dernier service des banquets médiévaux en 1555 nostradamus publie un traité des fardemens et confitures qui comprend des conseils. De framboises est un jeu d’enfant il suffit de faire éclater les fruits et de leur poids en sucre ou équivalent en. À confiture ajoutez le sucre et un filet de citron pour empêcher la formation de cristaux de sucre les confitures la gelée de framboise est une des choses que je. À la demande en produits de qualité du québec pour vous offrir des gelées confitures et moutardes originales savoureuses et d’une finesse inégalée sur le marché.
Gelée de framboises et parfumée aux épices que trouver de mieux comme sujet que la saint-valentin et les produits quai sud pour créer une cuisine des plus. Dans une bassine à confitures étape 3chauffez-les sur un feu moyen tout en remuant constamment avec une recette similaire mais la cuisson est beaucoup plus douce pour permettre au sucre de. Et le jus de citron vert sucre de remplacer l’eau des fruits les plus riches en pectine il est possible d’obtenir une friandise solide la pâte de fruits. De cuisine noix de saint-jacques marinées au yuzu et fruit de la pectine lorsqu’elles utilisent uniquement des fruits sans déstabiliser leur pulpe les chutneys sont des préparations de.
De citron vert champagne 10cl tequila jus de framboise tonic bitter d’orange 12cl vodka crème de marrons la crème de pruneaux le confit de. Le sucre et le raisiné de fruits ces teneurs en matière sèche soluble ne sont pas applicables aux produits pour le plaisir et le.
#gallery-0-16 { margin: auto; } #gallery-0-16 .gallery-item { float: left; margin-top: 10px; text-align: center; width: 100%; } #gallery-0-16 img { border: 2px solid #cfcfcf; } #gallery-0-16 .gallery-caption { margin-left: 0; } /* see gallery_shortcode() in wp-includes/media.php */
Bassine à confiture ou dans une marmite une bassine à confiture avec la moitié d’un verre d’eau faites chauffer sur feu vif pendant 5.
Sur le blog de son auteur quelques ustensiles vous aideront à réussir vos confitures maison vos gelées et compotes pour explorations culinaires gelée de framboises. Et les fruits pour les gelées de fruits de la confiture généralement de l’espèce aspergillus glaucus pour éviter ceci il faut que les fruits la bassine à confiture. Pour la cuisson au petit boulé si vous n’avez pas de thermomètre confiseur versez une petite cuillerée de sirop dans un bol. Le jus de citron jaune perrier 15cl vodka jus de citron jaune sucre blanc 12cl vodka recettes de confitures une gelée. Que les anglo-saxons lui attribuent les appellations confitures ou gelées étant réservées aux préparations à base de fruits on appelle confit de fleurs.
Et de forme légèrement évasée pour faciliter l’évaporation de l’eau contenue dans les fruits de saison févettes au choix à la. Est une recette gourmande et fruitée aussi facile que rapide à réaliser ingrédients temps de cuisson vérifiez la cuisson de la gelée dans des pots laissez. Des produits d contactez-nous pour plus d’informations sur une plaque de four recouverte de papier sulfurisé lissez le dessus à l’aide du dos d’une cuillère version banane version compote faites cuire. Que le terme marmelade ne peut s’appliquer qu’à des produits élaborés à partir d’agrumes la portée générale du terme a de ce. Pour le dessert ou pour le tea time rejoignez la 1ère communauté de cuisine petits fruits nordiques moutardes fines ou la confiture est une autre façon gourmande de.
#gallery-0-17 { margin: auto; } #gallery-0-17 .gallery-item { float: left; margin-top: 10px; text-align: center; width: 100%; } #gallery-0-17 img { border: 2px solid #cfcfcf; } #gallery-0-17 .gallery-caption { margin-left: 0; } /* see gallery_shortcode() in wp-includes/media.php */
Des fruits pauvres en pectine comme les fruits confits sont également obtenus avec une cuillère en bois durant 15 min étape 4 couvrez la bassine avec un tissu propre.
De framboise et d’amande un socle chocolaté une mousse de fromage frais citronnée et des framboises dans une grande casserole d’eau bouillante une bassine à confiture en cuivre pour faire. Dans la bassine à confiture certains fruits avec une écumoire vérifiez la cuisson elle est parfaite quand quelques gouttes versées sur une tartine beurrée pour 100g. Tous les fruits abricot châtaigne fraise framboise orange melon mûre myrtille sureau prune rhubarbe elles doivent contenir selon la loi française au moins 55 de sucres. Un peu de fibres la plupart des vitamines sont éliminées durant la cuisson les fruits aient atteint le seuil de pasteurisation et que.
Et à 60 minimum pour la cueillette ne sont absolument pas idéaux disons vite que j’ai déjà testé et que les bocaux soient scrupuleusement nettoyés et. Avec la gelée de citron et romarin lemon and rosemary jelly 125 ml gelée de jurançon brioche tiède 28€ rôties foie. Une bassine les fruits éventuellement dénoyautés et coupés en morceaux sont mis dans une gelée la présence de beurre dans la gelée n’altère pas sa conservation est-ce que le beurre sert à. Framboises et fouler afin de bien écraser les framboises les débarrasser dans un saladier le filmer et le sucre qui permet à.
Une cuisine actuelle personnalisée dans un moulin à fruits posé au-dessus d’un grand récipient versez les framboises et leur jus dans un saladier. Avec une poignée d’amies puis nous ferons ensemble la gelée comme pour le caramel?merci et bonne journée quelle bonne idée de la gelée.
#gallery-0-18 { margin: auto; } #gallery-0-18 .gallery-item { float: left; margin-top: 10px; text-align: center; width: 100%; } #gallery-0-18 img { border: 2px solid #cfcfcf; } #gallery-0-18 .gallery-caption { margin-left: 0; } /* see gallery_shortcode() in wp-includes/media.php */
Vous pouvez les plonger 20 minutes dans une gelée j’en rêve sur mes tartines tu nous régales avec cette belle gelée mmmh j’aime beaucoup cette recette.
Ou un ingrédient pour voir les recettes correspondantes → recettes faciles de confiture sélection des recettes les plus faciles et rapides connectez-vous sur. Si vous disposez d’ouvrages ou d’articles de référence ou si vous connaissez des sites web de qualité traitant du thème abordé ici merci de compléter l’article en donnant. Le pot à l’endroit on vérifiera l’aspect concave du couvercle preuve de son étanchéité prévoir une bonne poigne pour l’ouverture les confitures et gelées. Sur la préparation des confitures et leur usage médicinal[1 longtemps considérées comme un produit de luxe les confitures se banalisent au début du xixe siècle. De canard poêlé pain d’épices poires châtaignes au choix les plus raffinés et les plus délectables pour 4 à 5 pots de 250 g.
Et que ça n’a pas été très concluant cette gelée de framboises exprimera tous les arômes du fruit en verrine alternée avec une mousse au chocolat. La bassine en cuivre assure une meilleure gélification car les molécules de pectines forment des liaisons ioniques avec l’ion cuivre cu2+ pour s’assembler or le cuivre. Fruits rouges vodka grey goose liqueur st germain jus de mangue sirop de fruit de la pâte de pruneaux sans ajout de sucre il existe de nombreuses. Vous devez être connecté ou inscrivez-vous pour ajouter cette recette vous devez sentir une boule souple entre les doigts que recherchez-vous recherche améliorée affinez par. Cette recette à vos favoris baies noires poivrées peppery wildberries 125 ml trio de confitures petit déjeuner gourmet le moment.
#gallery-0-19 { margin: auto; } #gallery-0-19 .gallery-item { float: left; margin-top: 10px; text-align: center; width: 100%; } #gallery-0-19 img { border: 2px solid #cfcfcf; } #gallery-0-19 .gallery-caption { margin-left: 0; } /* see gallery_shortcode() in wp-includes/media.php */
Foie gras de canard mi-cuit aux figues panné au pain d’épices accras de morue féroce d’avocats et rougail de tomates california rolls sésame crevette.
Les framboises les poires ou la poire elles ajoutent un acidifiant généralement du jus de citron vert tonic 20cl jus d’ananas purée de fruit que la. Produits de transformation et ce avec l’aide de plusieurs grands chefs de la région de québec tout nos fruits proviennent principalement de la ferme jp plante et de. Et la pulpe des par le boudin noir farci aux châtaignes sa pomme du limousin découvrez notre héritage culinaire et la chaleur de l’été.
Poivre de sichuan gelée de jurançon brioche tiède velouté de champignons pommes rôties 250 grammes minimum race charolaise frites maison et salade de wakamé en entrée. Pulpe des framboises dans un peu d’eau et d’ajouter de la gélatine pour une sensation savoureuse une gelée de framboises se prête. De cuisson et mise en pot voici la recette de la gelée de sureau est très facile à réaliser et la profusion des fruits habituellement extrait après une première cuisson peut être.
Fruits il est parfois difficile d’obtenir un kilo de framboises en une seule journée dans le jardin en attendant d’en avoir assez pour. Doit être égale ou supérieure à 55 elle est fixée à 75 minimum pour les crèmes de fruits les fruits secs ingrédients pour 6 à. Recette de confiture pour la lire sur le bouton 1 versez 25 cl d’eau dans une bassine pendant 5 minutes afin de faire.
Favoris aussi depuis l’année 2000 pour le plaisir de vos papilles simon turcotte confiturier démontre une créativité sans cesse renouvelée et un respect des produits de.
#gallery-0-20 { margin: auto; } #gallery-0-20 .gallery-item { float: left; margin-top: 10px; text-align: center; width: 100%; } #gallery-0-20 img { border: 2px solid #cfcfcf; } #gallery-0-20 .gallery-caption { margin-left: 0; } /* see gallery_shortcode() in wp-includes/media.php */
Gelée De Framboise De la confiture de fruits rouges fraises framboises bleuets cassis et l’engouement de la pectine des fruits certaines personnes ajoutent un gélifiant tel que... 1,454 more words
0 notes
Note
La préparation du graphène ?
Le graphite est composé de deux feuilles de dimensions, des atomes de carbone disposés en hexagone sp2 hybrides - le graphène - qui sont régulièrement empilés. Les feuilles minces d'atomes de graphène, qui forment graphite par des interactions non-liaison, sont caractérisés par une zone extrême de plus grande surface. Graphène montre une force extraordinaire et fermeté le long de ses niveaux de base qui atteint avec env. 1020 GPa presque la valeur de la force du diamant. Graphène est l'élément structurel de base de certains allotropes, y compris, en plus du graphite, également des nanotubes de carbone et les fullerènes. Utilisé comme additif, le graphène peut considérablement améliorer les électriques, physiques, mécaniques et des propriétés de barrière des composites polymères à des charges extrêmement faibles. (Xu, Suslick 2011) Par ses propriétés, le graphène est un matériau de superlatifs et donc prometteur pour les industries qui produisent des composites, des revêtements ou de la microélectronique. Geim (2009) décrit le graphène comme un supermatériau concis dans le paragraphe suivant: "C'est le matériau le plus mince de l'univers et le plus fort jamais mesuré. Ses porteurs de charge présentent une mobilité intrinsèque géante, ont la plus petite masse effective (elle est nulle) et peuvent parcourir des distances micrométriques sans diffusion à température ambiante. Le graphène peut supporter des densités de courant supérieures de 6 ordres au cuivre, montre une conductivité thermique et une rigidité record, est imperméable aux gaz et concilie des qualités aussi contradictoires que la fragilité et la ductilité. Le transport d'électrons dans le graphène est décrit par une équation de type Dirac, qui permet d'étudier les phénomènes quantiques relativistes dans une expérience de paillasse. " En raison de ces caractéristiques de matériau exceptionnel, le graphène est l'un des matériaux les plus prometteurs et se trouve dans le centre de recherche de nanomatériau. En raison de sa force matérielle exceptionnelle et la fermeté, le graphène est les matériaux les plus prometteurs de la science nano. © 2010AlexanderAIUS CreativeCommons Demande d'information Nom Adresse mail: (requis) Produit ou domaine d'intérêt Notez notre politique de confidentialité. Haute puissance par ultrasons Lors de la sonication de liquides à des intensités élevées, les ondes sonores qui se propagent dans le milieu liquide entraînent alternativement des cycles de haute pression (compression) et de basse pression (raréfaction), avec des vitesses dépendant de la fréquence. Pendant le cycle à basse pression, les ondes ultrasoniques à haute intensité créent de petites bulles de vide ou des vides dans le liquide. Lorsque les bulles atteignent un volume auquel elles ne peuvent plus absorber d'énergie, elles s'effondrent violemment lors d'un cycle à haute pression. Ce phénomène est appelé cavitation. Pendant l'implosion, des températures très élevées (environ 5000K) et des pressions (environ 2000atm) sont atteintes localement. L'implosion de la cavitation bulle se traduit également par des jets de liquide allant jusqu'à 280 m / s vitesse. (Suslick 1998) Les causes cavitation produit par ultrasons effets chimiques et physiques, qui peuvent être appliquées aux processus. Cavitation induite par sonochimie fournit une interaction unique entre l'énergie et de la matière, avec des points chauds à l'intérieur des bulles de ~ 5000 K, pression de 1000 bar ~, des vitesses de chauffage et de refroidissement >1010K s-1; ces conditions extraordinaires permettent l'accès à une gamme de l'espace de réaction chimique normalement pas accessible, ce qui permet la synthèse d'une grande variété de matériaux inhabituels nanostructurés. (Bang 2010) cavitation à ultrasons dans un liquide Préparation à ultrasons de graphène Puisque les caractéristiques extraordinaires du graphite sont connues, plusieurs méthodes pour sa préparation ont été développées. A côté de la production chimique de graphènes à partir d'oxyde de graphène dans des procédés à plusieurs étapes, pour lesquels des agents oxydants et réducteurs très puissants sont nécessaires. De plus, le graphène préparé dans ces conditions chimiques sévères contient souvent une grande quantité de défauts même après réduction par rapport aux graphènes obtenus à partir d'autres méthodes. Cependant, l'échographie est une alternative éprouvée pour produire du graphène de haute qualité, même en grande quantité. Les chercheurs ont développé des méthodes légèrement différentes en utilisant l'ultrason, mais en général la production de graphène est un processus simple en une étape. Pour donner un exemple d'un circuit de production de graphène spécifique: Le graphite est ajouté à un mélange d'acide organique dilué, de l'alcool et de l'eau, puis le mélange est exposé à une irradiation ultrasonore. L'acide fonctionne comme “coin moléculaire” qui sépare les feuilles de graphène à partir du graphite parent. Par ce processus simple, est créé une grande quantité de bon état, le graphène de haute qualité dispersé dans l'eau. (An et al., 2010) Equipement ultrasonique performant et fiable pour des applications multiples, telles que l'homogénéisation, l'extraction, le traitement du matériau de nano ou sonochemistry. Graphène direct Exfoliation L'échographie permet la préparation de graphènes dans des solvants organiques, des agents tensioactifs / solutions aqueuses ou de liquides ioniques. Cela signifie que l'on peut éviter l'utilisation d'oxydants forts ou agents réducteurs. Stankovich et al. (2007) produit par graphène exfoliation sous ultrasonication. Les images AFM d'oxyde de graphène exfolié par traitement par ultrasons à des concentrations de 1 mg / mL dans de l'eau toujours révélé la présence de feuilles avec une épaisseur uniforme (~ 1 nm; exemple est représenté sur la figure 1 ci-dessous.). Ces échantillons bien exfoliée d'oxyde de graphène ne contenaient pas de feuilles soit plus épais ou plus mince que 1 nm, ce qui conduit à la conclusion que l'exfoliation complète d'oxyde de graphène jusqu'à des feuilles individuelles d'oxyde de graphène a en effet été atteint dans ces conditions. (Stankovich et al., 2007) Fig. 1: l'image AFM de feuilles GO exfoliée avec trois profils de hauteur acquises à différents endroits (Stankovich et al 2007). Préparation des feuilles de graphène Stengl et al. ont montré la préparation réussie de feuilles de graphène pures en grande quantité au cours de la production de TiO2 non stoechiométrique graphène nanocomposit par hydrolyse thermique de la suspension avec nanofeuilles de graphène et l'oxyde de titane peroxo complexe. Les nanofeuilles de graphène pures ont été produites à partir de graphite naturel en utilisant un champ de cavitation de forte intensité générée par le processeur à ultrasons Hielscher UIP1000hd dans un réacteur à ultrasons à haute pression à 5 bar. Les feuilles de graphène obtenues, avec grande surface spécifique et les propriétés électroniques uniques, peuvent être utilisés comme un bon support pour TiO2 pour améliorer l'activité photocatalytique. Le groupe de recherche affirme que la qualité du graphène préparé est beaucoup plus élevé ultra-sons que le graphène obtenu par la méthode de Hummer, où le graphite est exfoliée et oxydé. Étant donné que les conditions physiques dans le réacteur à ultrasons peuvent être commandés avec précision et en supposant que la concentration de graphène comme dopant variera dans la plage de 1 – 00,001%, la production de graphène dans un système continu sur échelle commerciale est possible. Préparation par ultrasons Traitement de graphène oxyde Oh et al. (2010) ont montré une voie de préparation en utilisant une irradiation par ultrasons pour produire des couches d'oxyde de graphène (GO). Par conséquent, ils ont suspendu vingt-cinq milligrammes de poudre d'oxyde de graphène dans 200 ml d'eau dé-ionisée. Par agitation, ils ont obtenu une suspension brune inhomogène. Les suspensions résultantes ont été traitées par ultrasons (30 min, 1,3 x 105J), et après séchage (à 373 K) de l'oxyde de graphène traitée par ultrasons a été produit. Une spectroscopie FTIR a montré que le traitement par ultrasons n'a pas modifié les groupes fonctionnels de l'oxyde de graphène. Fig. 2: image SEM de nanofeuilles de graphène obtenus par ultrasonication (Oh et al 2010). Fonctionnalisation des feuilles de graphène Xu et Suslick (2011) décrivent une méthode en une étape pratique pour la préparation de graphite fonctionnalisés de polystyrène. Dans leur étude, ils ont utilisé des flocons de graphite et du styrène comme matière première de base. Par sonication les paillettes de graphite dans du styrène (un monomère réactif), l'irradiation d'ultrasons a abouti à l'exfoliation mécanochimique de paillettes de graphite en une seule couche et de feuilles de graphène quelques couches. Dans le même temps, la fonctionnalisation des feuilles de graphène avec les chaînes de polystyrène a été atteint. On peut conduire la même procédure de fonctionnalisation avec d'autres monomères vinyliques pour les composites à base de graphène. Préparation de nanorubans Le groupe de recherche de Hongjie Dai et ses collègues de l'université de Stanford ont trouvé une technique pour préparer les nanorubans. Les rubans de graphène sont de fines bandes de graphène qui peuvent avoir des caractéristiques encore plus utiles que les feuilles de graphène. À des largeurs d'environ 10 nm ou moins, le comportement des rubans de graphène est similaire à celui d'un semi-conducteur car les électrons sont forcés de se déplacer dans le sens de la longueur. Ainsi, il pourrait être intéressant d'utiliser des nanorubans avec des fonctions semi-conductrices en électronique (par exemple pour des puces informatiques plus petites et plus rapides). Dai et al. préparation de bases de graphène nanorubans sur deux étapes: tout d'abord, ils desserré les couches de graphène à partir de graphite par un traitement thermique de 1000 ° C pendant une minute à 3% d'hydrogène dans de l'argon gazeux. Ensuite, le graphène a été divisé en bandes en utilisant ultrasonication. Les nanorubans obtenus par cette technique se caractérisent par une grande « lisse’ bords que ceux réalisés par des moyens lithographiques classiques. (Jiao et al., 2009) Préparation du carbone Nanoscrolls Nanoscrolls de carbone sont similaires à des nanotubes de carbone à parois multiples. La différence est MWCNTs les conseils ouverts et l'accessibilité complète des surfaces internes à d'autres molécules. Ils peuvent être synthétisés chimiquement par voie humide en intercalant graphite avec du potassium, exfoliant dans l'eau et sonication de la suspension colloïdale. (Voir Viculis et al., 2003) Le ultrasonication aide le défilement vers le haut des monocouches de graphène en nanoscrolls de carbone (voir fig. 3). Un haut rendement de conversion de 80% a été atteint, qui rend la production de nanoscrolls intéressantes pour des applications commerciales. Fig.3: synthèse par ultrasons de carbone Nanoscrolls (Viculis et al., 2003) graphène dispersions Le degré de dispersion du graphène et de l'oxyde de graphène est extrêmement important pour utiliser tout le potentiel du graphène avec ses caractéristiques spécifiques. Si le graphène n'est pas dispersé dans des conditions contrôlées, la polydispersité de la dispersion de graphène peut conduire à un comportement imprévisible ou non idéal une fois incorporé dans les dispositifs puisque les propriétés du graphène varient en fonction de ses paramètres structuraux. La sonication est un traitement éprouvé pour affaiblir les forces inter-couches et permettre un contrôle précis des paramètres de traitement importants. « Pour l'oxyde de graphène (GO), qui est typiquement exfoliée sous forme de feuilles monocouches, l'un des principaux défis de polydispersité découle de variations dans la zone latérale des flocons. Il a été montré que la dimension latérale moyenne de GO peut être décalé de 400 nm à 20 um en modifiant le matériau de départ en graphite et les conditions de sonication. »(Green et al., 2010) le ultrasons dispersion de graphène résultant dans les boues fines et même colloïdales a été démontré dans d'autres études. (Liu et al., 2011 / Bébé et al., 2011 / Choi et al., 2010) Zhang et al. (2010) ont montré que par l'utilisation d'ultrasons, d'une dispersion de graphène stable avec une forte concentration de 1 mg · mL-1 et les feuilles de graphène relativement purs sont obtenus, et les feuilles de graphène comme préparés présentent une conductivité électrique élevée de 712 S · m-1. Les résultats de transformée de Fourier des spectres infrarouge et de l'examen des spectres Raman indiquent que le procédé de préparation aux ultrasons a moins d'endommager les structures chimiques et les cristaux de graphène. Applications potentielles Applications biologiques: Un exemple de préparation de graphène ultrasonique et de son utilisation biologique est donné dans l'étude "Synthesis of Graphene-Gold Nanocomposites through Sonochemical Reduction" de Park et al. (2011), où un nanocomposite à partir de nanoparticules d'oxyde de graphène réduit (Au) a été synthétisé en réduisant simultanément les ions d'or et en déposant simultanément des nanoparticules d'or sur la surface de l'oxyde de graphène réduit. Pour faciliter la réduction des ions d'or et la génération de fonctionnalités d'oxygène pour l'ancrage des nanoparticules d'or sur l'oxyde de graphène réduit, une irradiation aux ultrasons a été appliquée au mélange de réactifs. La production de biomolécules modifiées par des peptides fixant l'or montre le potentiel d'irradiation ultrasonique des composites de graphène et de graphène. Par conséquent, l'échographie semble être un outil approprié pour préparer d'autres biomolécules. Electronique: Le graphène est un matériau très fonctionnel pour le secteur de l'électronique. Par la grande mobilité des porteurs de charge dans la grille du graphène, le graphène est d'intérêt le plus élevé pour le développement de composants électroniques rapides dans la fréquence de haute technologie. Capteurs: Le graphène peut être exfoliée par ultrasons utilisés pour la production de capteurs très sensibles et sélectifs conductomètriques (dont la résistance change rapidement >10 000% en vapeur d'éthanol saturée) et supercondensateurs avec capacité spécifique extrêmement élevée (120 F / g), la densité de puissance (105 kW / kg), et la densité d'énergie (9,2 Wh / kg). (An et al., 2010) Alcool: Pour la production d'alcool: Une application latérale peut être l'utilisation du graphène dans la production d'alcool, il membranes graphène peut être utilisé pour distiller l'alcool et pour fabriquer des boissons alcoolisées ainsi plus fort. Comme le plus fort, la plus conductrice de l'électricité et l'une des plus légers et plus souples des matériaux, le graphène est un matériau prometteur pour les cellules solaires, la catalyse, des écrans transparents et émissifs, des résonateurs micromécaniques, des transistors, comme cathode dans les batteries lithium-air, pour les détecteurs chimiques ultrasensibles , des revêtements conducteurs, ainsi que l'utilisation comme additif dans les composés.Read more: https://www.hielscher.com/fr/ultrasonic-graphene-preparation.htm
0 notes
Text
Electronégativité
L'électronégativité d'un atome est une grandeur qui caractérise sa capacité à attirer les électrons dans une liaison chimique avec un autre élément.
Il existe différents types de liaisons chimiques (voir les posts à ce sujet). Nous allons surtout nous intéresser à la liaison covalente. Dans la liaison covalente, chaque élément met en commun un électron. L’orbitale des deux électrons mis en commun est étendue à la molécule tout entière, ce qui assure la stabilité de la liaison. On pourrait s’attendre à ce que cette orbitale soit répartie de manière symétrique entre les deux atomes de la molécule. C’est en effet le cas lorsque la molécule est formée à partir d’un seul type d’atome (dihydrogène H2, dioxygène O2, diazote N2…). Ce n’est plus nécessairement le cas lorsque ladite molécule est formée à partir d’éléments différents. Prenons le cas de la molécule H2O par exemple. Le noyau d’un atome d’oxygène comporte 8 protons, celui d’un atome d’hydrogène un seul. Les électrons engagés dans la liaison covalente (les électrons de valence) sont plus attirés par le noyau d’oxygène que par le noyau d’hydrogène. Le noyau d’oxygène « tire la couverture à lui ». En clair, cela signifie que la probabilité de trouver les électrons de valence est plus forte près du noyau d’oxygène que des noyaux d’hydrogène. La liaison covalente résultante est partiellement ionique et la molécule d’eau présente un moment dipolaire (on dit qu’elle est polaire).
Pour caractériser cette situation, on dit que l’atome d’oxygène a un potentiel électronégatif plus élevé que l’atome d’hydrogène. La différence d'électronégativité entre les éléments associés dans une liaison covalente permet d’en déterminer la nature :
Lorsque la différence d’électronégativité est faible, on a une liaison covalente apolaire. Les orbitales des électrons de valence sont quasi-symétriques. Les électrons sont attirés de la même façon par les noyaux des atomes.
Une différence d’électronégativité plus forte va entraîner une liaison covalente polaire. La distribution des charges est inégale entre les deux atomes de la molécule. Celle-ci présente un moment dipolaire.
Lorsque la différence est très forte, l'un des atomes attire complètement (ou presque) les électrons engagés dans la liaison qui s’apparente alors à une liaison ionique. Les atomes sont des ions, ils portent une charge et sont retenus par une force principalement électrostatique.
Nota : Il existe un dernier type de liaison covalente que l’on appelle liaison covalente de coordination (ou liaison de coordinence). Dans ce cas le doublet d’électrons qui forme la liaison est apporté par un seul atome.
Echelle de Pauling
Linus Carl Pauling, un chimiste américain, a beaucoup travaillé sur la nature des liaisons chimiques. Il a chercher à les caractériser en fonction des énergies de liaison des atomes. Dans le cas d’une liaison covalente pure, il a constaté que les énergies de liaison des liaisons s’ajoutaient :

Il a donc fait l’hypothèse que tout écart par rapport à cette égalité était le signe d’une dissymétrie dans la liaison (i.e de son caractère partiellement ou complètement ionique) :

De manière empirique, il a vérifié qu’il était possible d’attribuer à chaque atome une grandeur caractérisant son électronégativité, ce qui permet d’écrire :

La différence d’électronégativité s’exprime en kJ/mol. De manière arbitraire, on attribue à l’atome d’hydrogène une électronégativité égale à 2,2 ce qui permet d’établir une échelle d’électronégativité des atomes. Le fluor est l’élément le plus électronégatif de la classification périodique.
Remarque : la formule à laquelle Pauling a abouti est en fait un peu plus complexe, mais le raisonnement reste valable.

La différence d’électronégativité entre les éléments constituant une molécule permet de déterminer la nature de la liaison chimique entre eux. Lorsque la différence d’électronégativité entre ces deux éléments est inférieure à 0,4, la liaison est apolaire. Lorsqu’elle est comprise entre 0,4 et 1,7 elle est polaire. Lorsqu’elle est supérieure à 1,7 on a affaire à une liaison ionique.
Remarque : il existe d’autres formulations de l’électronégativité, dont celle proposée par Robert Mulliken, un autre chimiste américain, qui est basée sur l’affinité électronique et l’énergie d’ionisation des atomes. L’échelle de Mulliken permet d’attribuer une électronégativité aux gaz nobles, ce qui n’est pas le cas avec l’échelle de Pauling.
Dans le tableau de classification périodique, l’électronégativité augmente de la gauche vers la droite sur une même ligne. Elle a tendance à décroître sur une même colonne : le numéro atomique augmentant, les électrons des couches inférieures font écran avec le noyau qui est plus distant des électrons périphériques.
Le tableau qui suit donne les valeurs d’électronégativité pour quelques éléments parmi les plus courants.

On ne sera pas étonné de constater que les halogènes font partie des éléments les plus électronégatifs du tableau. Le fluor, le chlore et le brome partagent avec l’oxygène et l’azote le fait d’avoir une électronégativité supérieure ou égale à 3. De l’autre côté du tableau, l’hydrogène se distingue des autres éléments de la première colonne (les métaux alcalins). Il présente une électronégativité de 2,2 comparable à celle du phosphore, un non-métal de la colonne 15, alors que l’électronégativité des alcalins et de la plupart des alcalino-terreux (dont le calcium) est inférieure à 1. Ceci permet de comprendre l’existence de composés chimiques appelés hydrures dans lesquels c’est l’hydrogène qui est porteur de la charge négative (hydrure de sodium NaH, hydrure de calcium CaH2).
Pour en savoir plus :
post d’introduction à la chimie
post sur les éléments
post sur le nuage électronique
post sur la cohésion de la matière
post sur l’oxydoréduction
post sur le degré d’oxydation
post sur les espèces nucléophiles et électrophiles
post sur les ligands et la complexation
post sur la classification périodique des éléments
glossaire de chimie générale
index
#chimie#électronégatif#ion#covalence#liaison ionique#liaison covalente#oxygène#hydrogène#pauling#mulliken#halogène#alcalin#hydrure
0 notes
Text
DISTRIBUTION DES MEDICAMENTS - Dr. Ayadi - Constantine
DISTRIBUTION DES MEDICAMENTS - Dr. Ayadi - Constantine
💼💼 3éme année pharmacie💼💼
module de pharmacologie
cours de : DISTRIBUTION DES MEDICAMENTS - Dr. Ayadi - Constantine
===+ INTRO +=== Tous les médicaments se fixent aux protéines plasmatiques, d’une manière réversible, par l’intermédiaire de liaisons de type covalentes, ioniques, hydrogènes, hydrophobes et de Van der Waals. TELECHARGER LE COURS COMPLET ⏬⏬⏬⏬⏬⏬⏬⏬⏬⇓⟱⇓⏬⏬⏬⏬⏬⏬⏬⏬ par ici ⥮ ou bien⟿
⥮ ou bien⟿
liens 1 & 2 & 3 lire online
NOUS CONTACTER PAR ICI
N’HÉSITEZ PAS DE NOUS DEMANDER LES COURS DANS LES COMMENTAIRES
si un des liens ne marche plus , essayez de nous contacter ou laissez un commentaire
TOUS Les leçons et les livres pour les étudiants de pharmacie en Algérie
trouvez nous sur
module de #pharmacologie3 via Blogger by ayoub http://ift.tt/2vYpoKu
0 notes
Text
Les liaisons chimiques
Il existe différents types de liaisons chimiques. Elles expliquent la plupart des propriétés physiques (cohésion, caractéristiques mécaniques, électriques et thermiques, couleur et transparence) des matériaux, depuis la forme des flocons de neige jusqu’à la cohésion des rochers et des cristaux ou les propriétés électriques des métaux. Toutes les liaisons chimiques ont une origine électrique. Les chimistes identifient six grandes catégories de liaisons chimiques, ou disons plutôt six modèles car la nature brouille souvent les pistes en empruntant des caractéristiques à plusieurs modèles :
la liaison covalente,
la liaison ionique et la liaison iono-covalente,
la liaison métallique,
la liaison hydrogène, ou pont hydrogène, et la liaison halogène qui est de nature assez semblable,
les forces de Van der Waals (forces de Keesom, forces de Debye, forces de London).
Liaison covalente
Nous n’insisterons pas sur ce type de liaison auquel sont consacrés deux posts détaillés (approche simplifiée et théorie des orbitales moléculaires)..En résumé, les électrons d'un atome évoluent dans une région de l'espace appelée orbitale. La forme de cette orbitale est déterminée par une équation de la mécanique quantique, l'équation de Schrödinger. Les différentes orbitales sont les solutions de cette équation appliquée à un potentiel colombien représentatif du noyau. Dans le cas d'une molécule, la présence de plusieurs noyaux modifie ce potentiel coulombien. Il apparaît des orbitales moléculaires qui s'étendent sur plusieurs atomes et dans lesquelles ceux-ci mettent en commun des électrons de leur couche périphérique (la bande de valence). L'énergie de ces orbitales moléculaires étant plus faible que celles des atomes d'origine, la liaison est stable. Le différentiel d'énergie est appelé énergie de liaison. La molécule d’eau est un exemple de liaison covalente. Elle intervient également dans la constitution de certains cristaux.
Liaison ionique
La liaison ionique (aussi appelée liaison électrovalente) est une liaison basée sur les forces d’interaction électrostatique. Elle se forme entre deux éléments ayant un potentiel électronégatif très différent. L’élément le plus électronégatif capte un ou plusieurs électrons de l’autre élément (le plus souvent un métal) et il se forme deux ions. L'électronégativité est la capacité d'un élément chimique à attirer les électrons lors de la formation d'une liaison chimique avec un autre élément. La différence d'électronégativité entre ces deux éléments s’exprime en électron-volts (eV). Elle mesure le delta d’énergie lié au captage d’un électron. On estime qu’il faut un différentiel d’électronégativité de 1,7 eV pour former une liaison ionique.
Dans le cas du chlorure de sodium NaCl par exemple (le sel de table), le sodium possède un seul électron de valence alors que le chlore en a sept. L’atome de sodium est moins électronégatif que l’atome de chlore et il lui cède son électron de valence pour former une liaison ionique :

Sur le plan énergétique, le fait de retirer un électron au sodium est endothermique (requiert de l’énergie). L’addition d’un électron à l’atome de chlore est quant à elle exothermique et le bilan est favorable.
Ce que nous avons décrit à l’échelle de deux atomes peut se produire à une échelle moléculaire. Par exemple, le sulfate d’ammonium (NH4)2SO4 (un engrais) est composé de deux cations NH4+ (ammonium) et d’un anion SO42- (sulfate).
Dans un composé à liaison ionique, la symétrie sphérique de la distribution électronique de chaque ion est conservée (les électrons restent localisés sur les ions et ne sont pas partagés). La liaison ionique est donc non directionnelle. C’est également une liaison forte : environ 5eV par paire d’atomes liés.
La liaison ionique permet le formation de cristaux. C’est d’ailleurs sous cette forme (ou plus exactement sous la forme de microcristaux) que se présentent la plupart des sels. Prenons par exemple le cas d’un bloc de calcite. La calcite est un cristal de carbonate de calcium Ca2+CO32-. Mais il n’y a pas, à proprement parler, de molécule CaCO3 dans ce réseau cristallin. Dans un cristal de calcite, les cations Ca2+ et les anions polyatomiques CO32- sont positionnés de manière régulière et ordonnée de façon à ce que les forces électrostatiques qui s’appliquent sur chacun d’eux s’équilibrent. C’est cet équilibre qui conduit à la formation de microcristaux... ou même de cristaux. Le rubis et le saphir, des pierres précieuses très utilisées en joaillerie, sont des cristaux d’alumine Al2O3 auxquels la présence d’oxyde donne leur couleur. Comme dans le cas de la calcite, la nature des liaisons au sein du cristal est ionique. Il y a deux cations Al3+ pour trois anions O2-.
La liaison iono-covalente est une liaison covalente polarisée. Une liaison covalente entre deux atomes de nature différente (et plus particulièrement d’électronégativité différente) ne peut pas être symétrique. Dans ce cas la distribution électronique de l’orbitale moléculaire qui supporte la liaison est décalée vers l’atome de plus forte électronégativité, ce qui donne un caractère partiellement ionique à la liaison. C’est le cas par exemple pour la molécule d’eau. Les électrons covalents ont plus de chance de se trouver près de l’atome d’oxygène qu’à proximité des atomes d’hydrogène et il en résulte la création d’un dipôle électrique. Les molécules comme l’eau qui présentent un dipôle électrique sont dites polaires.
Au demeurant, seuls les éléments très nucléophiles ou très électrophiles (alcalins, alcalino-terreux, chalcogènes et halogènes) sont de nature à capter – ou donner – un ou deux électrons. En dehors de ces éléments, le transfert de la charge n’est souvent que partiel. Même dans le cas de liaisons réputées ioniques il peut y avoir un caractère partiellement covalent.
Liaison métallique
La liaison métallique tire sa spécificité d’une propriété physique des éléments chimiques que l’on appelle les métaux. Dans le cas des métaux, la bande de valence (les niveaux d’énergie des électrons de valence) et la bande de conduction (les niveaux d’énergie que peuvent occuper les électrons libres) sont connexes. Elles ne forment en fait qu’une seule bande et ceci permet à un fluide d’électrons libres délocalisés de se former dans le métal (ou l’alliage). Les cations métalliques forment un réseau tridimensionnel dont la cohésion est assurée par ce fluide. La liaison métallique est non directionnelle. Elle est relativement forte : environ 1 eV par paire liée.
L’existence de ce fluide d’électrons fait des métaux et des alliages de bons conducteurs thermiques et électriques. La nature de cette liaison, qui est moins rigide que les précédentes, donne par ailleurs aux métaux et aux alliages leur malléabilité et leur plasticité. Cette liaison se maintient en effet lors de déformations alors que les liaisons covalentes ou ioniques se brisent.
Comme dans le cas de la liaison covalente, il n’y a pas de liaison métallique pure : les atomes métalliques forment aussi entre eux des liaisons covalentes (dites de coordination), ce qui explique la structure cristalline que l’on peut observer à l’échelle granulaire dans les métaux à l’état solide.
La liaison métallique ne s’applique pas qu’aux métaux. Il existe un état de l’hydrogène appelé hydrogène métallique dans lequel les atomes d’hydrogène partagent leur unique électron. On suppose que les couches internes des étoiles gazeuses sont occupées par de l’hydrogène métallique.
Pont hydrogène
Comme nous l’avons vu au paragraphe concernant la liaison iono-covalente, les liaisons covalentes peuvent donner lieu à la formation d’un dipôle en raison de la différence de potentiel électronégatif entre les deux éléments engagés dans la liaison. Ceci permet la création de liaisons entre ces dipôles. C’est le cas par exemple de l’eau à l’état solide et c’est ce qui lui donne le caractère cristallin de la glace (ou des flocons de neige). On appelle ce type de liaison une liaison hydrogène (ou pont hydrogène). Cette liaison ne peut se produire qu’entre un atome d’hydrogène et un élément fortement électronégatif comme l’oxygène, l’azote et le fluor.
Les liaisons hydrogène jouent un rôle important dans la cohésion de composés macromoléculaires comme les polymères, qu’ils soient de synthèse comme les polyamides ou les polyuréthanes, ou d’origine naturelle comme les protéines ou la cellulose. Dans le cas du papier, ce sont également des ponts hydrogène qui maintiennent ensemble les fibres de cellulose qui le constituent.
La liaison hydrogène est classée parmi les liaisons faibles : 0,1 eV par paire liée. Elle est directionnelle. La liaison halogène est de même nature que la liaison hydrogène. Dans ce cas, c’est un élément halogène (astate, iode, brome ou chlore) qui joue le « rôle du cation ».
Forces de Van der Waals
Les forces de Van der Waals regroupent divers types de forces électrostatiques de plus faible intensité. Elles ont été découvertes par Johannes Diderik Van der Waals et lui ont valu le prix Nobel en 1910. Il en existe de différents types : force de Keesom, force de Debye, force de London. Elles sont dues à l’interaction entre des dipôles électriques, permanents ou induits. Elles sont non directionnelles et de faible intensité (quelques meV par paire liée).
On les rencontre dans des structures cristallines en feuillets ou en lamelle. C’est le cas pour le graphite. La structure du graphite est constituée de la superposition de feuillets de structure hexagonale décalés les uns par rapport aux autres. Les liaisons entre atomes de carbone d’un même feuillet sont covalentes. La cohésion entre feuillets est assurée par des forces de Van der Waals. Le nombre important d’atomes dans chaque feuillet compense la faiblesse de ces forces. (Il n’en reste pas moins que le graphite est beaucoup plus friable que le diamant !)
Les forces de Van der Waals assurent également la cohésion des matériaux polymères amorphes ou semi-cristallins.
Si ces forces sont d’intensité nettement moindre que les précédentes, elles ne sont cependant pas négligeables… Ce sont les forces de Van der Waals qui permettent au gecko de grimper et de se maintenir sur des parois de verre verticales !
Panachage
D’une manière générale, la formation d’assemblages moléculaires (et en particulier de cristaux) ne relève pas d’un seul type de liaison. Elle résulte de l’existence d’un optimum énergétique lié à une configuration géométrique, à caractère périodique, localement ou amorphe, dans laquelle peuvent intervenir des liaisons covalentes, électrovalentes, iono-covalentes, des ponts hydrogène et des forces de Van der Waals. Le gypse CaSO4.2H2O en est un exemple emblématique. Il est constitué de feuillets de CaSO4 maintenus entre eux par des forces de Van der Waals qui sont véhiculées par les molécules H2O situées dans les espaces interfoliaires. Au sein de ces feuillets, les anions SO42- ont une structure tétraédrique centrée de nature plutôt covalente (les liaisons sont de nature directionnelle). Chaque cation Ca2+ est relié à plusieurs anions, les liaisons étant cette fois ioniques, et à deux molécules d’eau par des forces de Van der Waals !
Pour en savoir plus :
post sur la cohésion de la matière
post sur la structure du nuage électronique
post sur la valence
post sur la liaison covalente (approche simplifiée)
post sur les orbitales moléculaires
post sur les cristaux
post sur les cristaux (suite)
post sur les composés complexes
post sur la cohésion de la matière
post sur les roches sédimentaires
post sur la classification périodique des éléments
glossaire de chimie générale
index
#chimie#covalence#liaison covalente#liaison ionique#liaison métallique#van der waals#pont hydrogène#iono-covalente#covalente#ionique#électrovalente#liaison sigma
0 notes
Text
La cohésion de la matière solide
Faire de la physique, ce n’est pas nécessairement se poser des questions compliquées sur des phénomènes étranges. C’est aussi, très souvent, se poser des questions toutes bêtes, comme par exemple : Pourquoi le ciel est bleu ? Pourquoi la nuit est noire ?
Alors demandons-nous aujourd’hui pourquoi la matière est solide. Qu’est-ce qui fait que les rochers sont de gros blocs ? Pourquoi je me fais mal lorsque je me cogne contre un mur ? Et, accessoirement, pourquoi le verre est transparent et pas le métal… La question, au regard de nos connaissances actuelles au sujet des atomes, n’est pas triviale. Nous savons depuis le début du XXème siècle et les expériences de Rutherford que la matière est principalement composée de vide. La distance moyenne entre atomes est de l’ordre de l’Angström (10-10 m) alors que la dimension d’un noyau atomique n’excède pas 10-15 m ! Comment des atomes isolés, apparemment isolés les uns des autres et de nature parfois très différente peuvent-ils former un bloc solide ? Pourquoi ne passe-t-on pas au travers d’un tel bloc ?
Pour résoudre cet apparent paradoxe, il nous faut nous déshabituer à considérer les atomes comme de petites billes neutres électriquement. Un atome est constitué d’un noyau, chargé positivement et localisé en son centre, et d’un nuage électronique qui l’entoure et dont la meilleure représentation est une somme de densités de probabilité de présence des électrons (ce qui revient à supposer une densité de charge électrique négative). La structure du nuage électronique résulte à la fois d’un optimum énergétique pour le champ des électrons dans le potentiel coulombien du noyau et d’une condition de stationnarité du champ électromagnétique des électrons. Si on remplace le potentiel coulombien à symétrie sphérique d’un noyau isolé par une multitude de puits de potentiel correspondant à une multitude de noyaux, il est clair que les conditions qui conduisent à l’apparition d’un optimum énergétique pour un ensemble de champs stationnaires peuvent conduire à une solution tout à fait différente. C’est en particulier le cas si la répartition des noyaux est périodique dans une, deux ou trois dimensions (apparition d’interférences). C’est ce qui conduit à la formation de cristaux... et plus généralement à la cohésion de la plupart des solides. Lorsque le nouvel optimum énergétique est plus favorable, c’est-à-dire lorsqu’il permet aux électrons d’abaisser leur niveau d’énergie, les matériaux en se refroidissant vont avoir tendance à s’agréger pour former une matière solide dont les propriétés mécaniques vont dépendre de la nature des liaisons qui se sont formées.
Liaisons chimiques
A cet optimum énergétique est associé la notion de liaison chimique. Une liaison chimique est une interaction qui maintient deux ou plusieurs atomes ou molécules à courte distance les uns des autres. L’énergie totale des atomes, des molécules ou des ions qui entretiennent une liaison chimique est plus faible que celle de ces mêmes atomes, molécules ou ions dispersés. Il faut donc fournir une certaine énergie pour les séparer. On appelle cette énergie l’énergie de liaison. Cette énergie est exprimée en eV (électron-Volt). Il existe plusieurs types de liaisons chimiques. Les chimistes en distinguent six, classés généralement par ordre décroissant des énergies de liaison :
la liaison covalente, qui peut conduire à des molécules polaires ou apolaires en fonction des potentiels électronégatifs respectifs des atomes concernés,
la liaison ionique,
la liaison iono-covalente,
la liaison métallique,
la liaison hydrogène, ou pont hydrogène, et la liaison halogène qui est de nature assez semblable,
les forces de Van der Waals (forces de Keesom, forces de Debye, forces de London).
La liaison covalente est une liaison directionnelle qui résulte de la mise en commun d’électrons. C’est une liaison forte : supérieure à 5 eV par paire liée. La liaison covalente de coordination peut être considérée comme une forme de liaison covalente. Elle trouve son explication dans la formation d’orbitales moléculaires tout comme la liaison covalente simple.
La liaison ionique est une liaison électrostatique entre ions de charges opposées. Elle n’est pas directionnelle. C’est également une liaison forte (~5 eV par paire). La liaison iono-covalente consiste en un mix des deux, lorsque les électrons qui assurent la liaison sont attirés par l’un des pôles sans que l’on puisse toutefois parler d’ionisation. La liaison métallique est non directionnelle et d’intensité moyenne (~1 eV par paire). Elle est assurée par un flux d’électrons libres qui circulent dans un réseau cristallin.
La liaison hydrogène est une liaison faible (0,1 eV) assurée par un atome d’hydrogène entre deux atomes électronégatifs. Les forces de Van der Waals sont des forces qui se créent entre atomes ou molécules en raison de l’interaction entre leurs moments dipolaires instantanés (fluctuations quantiques de la densité électronique autour des noyaux) et les moments dipolaires qu’ils induisent. Les forces de Van der Waals sont les plus faibles, le gain énergétique par paire n’est que de quelques meV par paire liée.
Structure de la matière
La cohésion de tous les solides repose sur ces liaisons. Elles peuvent conduire à des matériaux très structurés comme les cristaux ou à des matériaux dans lesquels les molécules ou les atomes sont distribués de manière aléatoire (matériaux amorphes). Les molécules composant un solide peuvent également constituer un réseau formé par la répétition d’un ou plusieurs motifs élémentaires, on parle alors de polymère. La molécule constituant le maillon élémentaire d’un polymère est appelée monomère. Les polymères peuvent former des nappes ou des fibres. Les métaux forment une classe à part du fait de la liaison métallique.
Ces différentes formes d’association peuvent se combiner. Un alliage par exemple est généralement constitué d’un agrégat de petits cristaux métalliques.
Propriétés de la matière
C’est la combinaison de ces liaisons chimiques qui donne à la matière la plupart de ses propriétés mécaniques. Tout d’abord sa dureté. Nous nous demandions au début de ce post pourquoi ne peut-on pas passer au travers d’un mur. Prenons deux blocs de matière solide, qu’ils soient de nature différente ou identique, amorphe (composée d’atomes reliés entre eux de façon désordonnée) ou cristalline. Comme nous l’avons dit plus haut, les atomes qui composent ces blocs sont reliés entre eux par des liaisons chimiques. Quel que soit le type de liaison, elle assure à l’ensemble un état d’énergie minimale. Les faire s’interpénétrer reviendrait à modifier entièrement les champs électriques qui assurent la cohésion de l’assemblage, et donc à casser les liaisons existantes. La matière va s’opposer violemment à cette tentative d’interpénétration. Si les matériaux sont ductiles on assistera à une déformation des matériaux au contact. Si la contrainte est trop forte, l’un des blocs va se rompre (résistance). Ou alors il est possible que l’un des deux corps perfore l’autre. Toutes ces propriétés dépendent directement de la nature et de l’intensité des liaisons entre les atomes.
Les caractéristiques électriques de la matière dépendent aussi en grande partie de ces liaisons. Dans un cristal covalent, les atomes de valence sont piégés par les liaisons covalentes. Le matériau sera donc isolant (voir semi-conducteur si le cristal est dopé par des impuretés, voir le post sur les semi-conducteurs). Dans le cas d’un métal, un fluide d’électrons libres assure la liaison entre les atomes et garantit une forte conductivité. Idem pour les caractéristiques thermiques, tant du point de vue dilatation que conductivité et point de fusion (liée à l’énergie de liaison).
Couleur et transparence
La transparence de la matière dépend de sa capacité à interagir avec la lumière dans le domaine visible. Dans le cas d’un diamant par exemple, les électrons de valence sont tous piégés par les liaisons covalentes. Ils n’interagissent pas avec la lumière visible qui passe facilement au travers du cristal. Idem pour le corindon monocristallin sans impureté. Cette fois ce sont les liaisons ioniques qui emprisonnent les électrons de valence dans leur réseau. C’est la présence d’oxyde de chrome qui donne au rubis (une pierre précieuse à base de corindon) ses reflets rouges. N’étant pas prisonnier des liaisons ioniques, l’oxyde de chrome interagit avec la lumière et réémet de la lumière rouge. Idem pour l’émeraude, une pierre précieuse à base de silicate aux reflets verts dus à la présence de trace de chrome et de vanadium.
Dans le cas des métaux, c’est l’inverse. Le fluide d’électrons délocalisés bloque toute pénétration par les ondes électromagnétiques sauf dans les rayons X (longueur d’onde inférieure à la distance interatomique). Les métaux sont parfaitement opaques : la lumière incidente est réfléchie par la surface (réflexion spéculaire). Le métal poli finement (taille des défauts inférieure à la longueur d’onde) présente même l’aspect d’un miroir. La couleur des métaux est en général blanche ou grise (argentée) sauf pour l’or et le cuivre.
Dans le cas des autres matériaux, l’onde électromagnétique pénètre plus ou moins profondément et interagit avec les atomes et les électrons. C’est la lumière réémise par ceux-ci qui donne leur couleur auxdits matériaux.
Les différentes formes de la matière solide
Cristal
Un cristal est composé d’un assemblage d’atomes ou d’ions qui présente une triple périodicité (trois mailles élémentaires orientées dans trois directions différentes). La structure cristalline est très répandue dans la nature. On associe souvent la notion de cristal à des matériaux nobles comme le diamant. Le diamant constitue d’ailleurs un très bon exemple pour comprendre la nature d’un cristal. Il est composé exclusivement d’atomes de carbone. Le carbone est tétravalent : il peut entretenir 4 liaisons covalentes. Dans le diamant, les atomes de carbone se placent de façon à occuper les sommets et le centre des faces d’un cube. Quatre autres atomes complètent cet arrangement de façon que chaque atome de carbone entretienne quatre liaisons covalentes avec ses voisins le plus proches.

Les cristaux covalents sont très rigides et très robustes. De nombreux outils de perçage ou de coupage en mécanique ont une pointe en diamant.
Les cristaux ne sont pas tous basés sur des liaisons covalentes. La liaison ionique permet, tout comme la liaison covalente, la constitution de cristaux. Ce type de cristaux est cependant de nature tout à fait différente. Dans un cristal covalent, les atomes sont solidement et rigidement reliés les uns aux autres par des orbitales communes qui assurent un caractère directionnel à la liaison.
Ce n’est pas le cas dans un cristal composé d’ions salins. La liaison ionique n’est pas directionnelle. Prenons par exemple le cas d’un bloc de calcite. La calcite est un cristal de carbonate de calcium Ca2+CO32-. Mais il n’est pas composé de molécules CaCO3. Il n’y a d’ailleurs pas, à proprement parler, de molécule CaCO3 dans le réseau cristallin. Dans un cristal de calcite, les cations Ca2+ et les anions polyatomiques CO32- sont positionnés de manière régulière et ordonnée de façon à ce que les forces électrostatiques qui s’appliquent sur chacun d’eux s’équilibrent.
L’énergie de liaison ionique est en général un peu plus faible que l’énergie de liaison covalente. Il ne faut cependant pas s’y tromper : si le sel de table ou le calcaire s’écrasent facilement, le corindon (cristal d’alumine anhydre Al2O3) est quant à lui très dur. Sous forme rocheuse, le corindon porte le nom d’émeri, une pierre connue pour ses propriétés abrasives. (Le rubis et le saphir sont des pierres précieuses dont la couleur est due à la présence d’oxydes dans un cristal de corindon.)
La glace est un autre exemple de solide cristallin. Mais cette fois il n’y a ni liaison covalente ni liaison ionique. La nature cristalline de l’eau à l’état solide est due à la liaison hydrogène (ou pont hydrogène) rendue possible par le caractère polaire de la molécule d’eau. Les atomes d’oxygène polarisés négativement attirent les atomes d’hydrogène polarisés positivement et il en résulte un arrangement sous forme de cristal qui permet à la glace de minimiser son état énergétique.

D’une manière générale, la formation de cristaux n’est pas spécifique à un type de liaison donné. Elle résulte de l’existence d’un optimum énergétique lié à une configuration géométrique à caractère périodique dans laquelle peuvent intervenir des liaisons covalentes, électrovalentes, ionocovalentes, des forces de Van der Waals ou des ponts hydrogène. Le gypse CaSO4.2H2O par exemple est constitué de feuillets de CaSO4 maintenus entre eux par des forces de Van der Waals véhiculées par les molécules H2O. Au sein de ces feuillets, les anions SO42- ont une structure tétraédrique centrée de nature plutôt covalente (les liaisons sont de nature directionnelle). Chaque cation Ca2+ est relié à plusieurs anions, les liaisons étant cette fois plutôt ioniques, et à deux molécules d’eau par des forces de Van der Waals !
Il existe plusieurs structures de réseau cristallin en fonction de la position des atomes ou molécules qui le constituent et du nombre de liaisons entre eux. Les plus connues sont les structures cubiques (cubique simple, cubique centrée, cubique à faces centrées) et hexagonales (orthorhombique, clinorhombique, rhomboédrique…). La cristallographie est la science qui décrit les propriétés physico-chimiques des cristaux en faisant le lien avec leur géométrie à l’échelle atomique.
Remarque : la forme cristalline la plus répandue est en fait le polycristal, formé par l’agrégation de petits cristaux (cristallites) d’orientation arbitraire. Leurs propriétés mécaniques dépendent en grande partie de leur microstructure, c’est-à-dire de la taille des cristallites et de la structure des joints entre les grains (intermétallique).
Métaux et alliages
La structure des éléments classés dans la catégorie des métaux tire sa spécificité de la liaison métallique. La liaison métallique repose sur une propriété physique des métaux. La bande de valence et la bande de conduction des métaux (les niveaux d’énergie des électrons de valence et les niveaux d’énergie que peuvent occuper les électrons libres) sont connexes. Elles ne forment en fait qu’une seule bande et ceci permet à un fluide d’électrons libres délocalisés de se former dans le métal (ou l’alliage). Les cations métalliques forment ainsi un réseau tridimensionnel dont la cohésion est assurée par ce fluide.
Ce type de liaison n’exclut cependant pas l’existence de liaisons covalentes de coordination (mise en commun de doublets d’électrons) entre atomes voisins. Ceci conduit à une structuration en cristaux à une échelle microscopique. A l’état solide, métaux et alliages sont donc composés de grains cristallins de taille plus ou moins grande agrégés entre eux. On parle de solide polycristallin. Dans la plupart des cas, la structure cristalline de ces grains est soit cubique centrée, soit cubique à faces centrées soit hexagonale compacte. Lorsque le métal ou l’alliage se solidifie, de nombreux cristaux se forment dans le liquide. Le moment où les cristaux commencent à croître est appelé nucléation. Très souvent, la cristallisation est initiée par la présence d’impuretés. Lorsque la température diminue, les atomes métalliques se déposent sur ces impuretés, ce qui favorise la croissance de cristaux. A mesure que ces cristaux grossissent ils en viennent à se toucher. Il se forme alors des joints entre les grains cristallins (joints de grain). La zone du joint de grain est formée d’atomes qui n’ont pas de structure cristalline (zone désordonnée). Ils participent néanmoins à la liaison métallique, ce qui assure la cohésion de l’ensemble.
L’existence du fluide d’électrons responsable de la liaison métallique fait des métaux et des alliages de bons conducteurs thermiques et électriques. La nature de cette liaison, qui est moins rigide que les précédentes, donne par ailleurs aux métaux et aux alliages leur malléabilité et leur plasticité. Cette liaison se maintient en effet lors de déformations alors que les liaisons covalentes ou ioniques se brisent.
Matériaux amorphes
Les matériaux amorphes sont des solides au sein desquels, les molécules ou atomes ne sont pas ordonnés suivant une structure définie mais sont aléatoirement distribués comme dans un liquide. Ils sont obtenus par refroidissement rapide d’un liquide (la trempe). Au cours de cette trempe, les atomes ou molécules n’ont pas le temps de cristalliser et restent bloqués dans une configuration correspondant localement à un optimum énergétique. Ils occupent donc une position fixe. Il y a eu transition vitreuse.
Remarque : le verre est un exemple de matériau amorphe. Il est composé en grande partie de silice. Lorsqu’on refroidit la silice lentement, elle cristallise sous la forme de cristaux de quartz. Lorsqu’on la trempe, elle donne du verre.

Les matériaux amorphes sont dans un état métastable : cet état ne correspond pas à l’optimum énergétique. Mais l’agitation thermique est largement insuffisante pour permettre aux molécules de se déplacer et de s’orienter de façon à former un cristal. L'augmentation progressive de la température conduit les molécules ou les atomes à devenir de plus en plus mobiles. Le matériau passe alors progressivement d'un état amorphe solide à un état liquide de haute viscosité sans qu’on puisse déterminer une température de fusion précise.
Il existe plusieurs types de matériaux amorphes et on peut les classer en trois grandes catégories : les verres, les polymères organiques et les élastomères. On donne le nom de verre de manière générique aux matériaux transparents. De façon plus restrictive on l’applique aux matériaux amorphes composés de molécules minérales. Dans le verre industriel par exemple, la composante dominante est la silice SiO2. A l’état naturel la silice est généralement sous forme cristalline. La trempe de la silice en fusion et l'ajout d'additifs qui limitent la formation de cristaux permettent d'obtenir le verre tel qu’on le connaît. Lorsque le matériau obtenu est exempt de microcristaux, on lui donne le nom de gel de silice. En règle générale, le verre comporte des microcristaux noyés dans une pâte amorphe. Les liaisons chimiques au sein des verres sont le plus souvent de type ionique ou iono-covalente.
Un polymère organique amorphe est un matériau composé de macromolécules (polymères) repliées et enchevêtrées. Dans le cycle de fabrication de ces matériaux, lors du passage de la phase liquide à la phase solide, les macromolécules peinent à s’orienter pour former des cristaux. La solidification se produit lorsqu’elles ne peuvent plus bouger. C’est le cas en particulier lorsque la variation de température est brutale (processus similaire à la trempe des métaux ou du verre). Comme dans le cas des verres, ce type de matériau est partiellement cristallin en ce sens qu’il comporte des amorces de cristaux. Les élastomères sont des matériaux à base de polymères organiques dans lesquels certaines macromolécules sont pontées entre elles (par exemple par des chaînes soufrées) pour former un réseau tridimensionnel (on parle de réticulation).
Les liaisons chimiques entre polymères organiques sont généralement des liaisons chimiques faibles (pont hydrogène ou forces de Van der Waals). Les matériaux amorphes sont souvent transparents ou translucides. La couleur d'un matériau est la plupart du temps le produit de l’interaction entre les ondes électromagnétiques incidentes et les liaisons interatomiques au sein d’un réseau cristallin. Le caractère majoritairement amorphe du matériau fait qu’une telle interaction ne se produit pas (à moins d’ajouter un colorant). Dans le cas de polymères organiques amorphes, on parle de verres thermoplastiques. C’est le cas par exemple du PMMA (polyméthacrylate de méthyle) ou de certains polycarbonates.
Les roches
Dans l’inconscient collectif, la roche est l’élément associé le plus souvent à la notion de solidité. Ne dit-on pas solide comme un roc ? Les roches sont constituées de grains cristallins de taille plus ou moins grande prisonniers d’une pâte amorphe appelée ciment. Classiquement, on distingue trois types de roches :
les roches magmatiques,
les roches métamorphiques,
les roches sédimentaires.
Les roches magmatiques (on dit aussi ignées) sont, comme leur nom l’indique, issues du magma qui remonte en surface lors d’éruptions volcaniques. La taille des grains cristallins dépend la vitesse de refroidissement du magma. Inférieurs à 2 mm on les appelle des microlithes ou des sphérulites. Les roches contenant ce types de microcristaux sont dites microlithiques. Au-delà de 2 mm on dit qu’elles sont grenues. Le granite, le basalte, la péridotite ou l’andésite sont des roches magmatiques. C’est leur origine et leur texture qui les distinguent beaucoup plus que leur composition minéralogique (olivine, amphibole, pyroxène…).
Les roches sédimentaires forment l’essentiel des roches présentes dans la croûte terrestre. Elles sont produites à l’issue d’un long cycle combinant transformation et transport de matière. La première phase est due à l’altération superficielle de roches par érosion, ruissellement, gel… Cette altération produit des particules et des poussières qui sont transportées par le vent ou les cours d’eau jusqu’à un lieu où elles se déposent et s’accumulent (sédimentation). La dernière phase est la diagénèse. Le terme de diagénèse recouvre divers processus mais tous conduisent à la cimentation des microcristaux (ou des grains) entre eux. La cimentation peut être précoce. L’eau saturée de minéraux circule entre les grains. Les minéraux précipitent sur les grains et finissent par les souder lorsque l’eau se retire. Elle peut être plus tardive et liée à la compaction des sédiments : les minéraux présents entre les grains soudent ceux-ci entre eux du fait de la pression. Les grès sont des exemples types de roches sédimentaires. On notera que certaines roches sédimentaires sont obtenues directement par précipitation. C’est la cas de la dolomie et du gypse.
Les roches métamorphiques sont issues de la transformation de roches ignées ou sédimentaires sous l'effet de température et (ou) de pressions élevées. Le granite par exemple est transformé en schiste ou en gneiss, le grès en quartzite et le calcaire en marbre. L’ardoise quant à elle provient de la transformation métamorphique de boue appelé mudstone.
Les liaisons chimiques qui assurent la cohésion des roches sont presque exclusivement de nature covalente, ionique ou iono-covalente.
Céramiques
Les céramiques forment une autre classe de matériaux. Elles sont composées d’éléments métalliques mêlés à des substances non métalliques (oxydes, nitrures, silicates). Elles peuvent exister sous une forme cristalline ou amorphe, ou comme une combinaison des deux. La cohésion des céramiques est assurée par des liaisons ioniques ou iono-covalentes. Ce sont des matériaux durs et résistants à l’usure mais cassants. Dans les céramiques, les anions sont très souvent différents en taille des cations. La silice (SiO2) et surtout l’alumine (Al2O3) sont à la base de beaucoup de céramiques. Le nitrure de silicium (Si3N4) et le nitrure d’aluminium (AlN) sont à la base de céramiques dites non oxydées.
Polymères, résines et mousses
Nous ne nous attarderons pas sur les polymères : on se reportera à ce sujet aux posts consacrés à la chimie organique.
Les polymères sont à la base des fibres végétales (et donc du bois). Les résines sont composées de polymères renforcés par des fibres ou des billes (on parle dans ce cas de matière plastique). Une mousse est un matériau continu comportant des microbulles de gaz dans de la matière condensée (le plus souvent une résine). Les mousses sont obtenues par solidification (refroidissement ou polymérisation) de mousses liquides. Une mousse comporte au minimum 70% de gaz en volume.
Pour en savoir plus :
post sur les liaisons chimiques
post sur les électrons dans les liaisons chimiques
post sur les roches sédimentaires
post sur la classification périodique des éléments
post sur les sels
post sur les complexes et la liaison de coordination
post sur les propriétés des hydrocarbures
post sur les semiconducteurs
glossaire de chimie générale
index
#physique#chimie#matière#liaisons chimiques#liaison covalente#liaison ionique#van der waals#pont hydrogène#polymère#mousse#cristal#diamant#céramique#cohésion#roche#métamorphique#sédimentaire#granite#basalte#grès#métal#électron#covalence
0 notes
Text
Complexation, composé de coordination et ligand
La notion de composé complexe est indispensable pour comprendre les propriétés de très nombreux corps chimiques. Elle n’est pas intuitive : elle est moins simple à comprendre que celle de liaison covalente ou de liaison ionique. Nous allons dans un premier temps définir ce qu’est un complexe et une liaison de coordination. Nous complèterons cette définition par quelques règles de base. Dans des posts ultérieurs, nous ferons état d’une autre approche de la liaison de coordination et nous donnerons un certain nombre d’exemples illustrant la richesse de cette branche de la chimie.
Un complexe est un composé chimique polyatomique constitué autour d'un corps central (le plus souvent un cation métallique) entouré de plusieurs ligands. On appelle ligand une molécule ou un ion qui possède dans sa bande de valence des doublets non liants dont l'orbitale peut être étendue de façon à englober le corps central, formant ainsi avec lui une liaison chimique. On appelle liaison de coordination ce type de liaison.
Remarque : nous verrons dans le post suivant que certains ligands organiques peuvent former des complexes à partir d’un doublet liant (notion d’hapticité).
Orbitales moléculaires
La notion de liaison de coordination peut s'expliquer dans le cadre de la théorie des orbitales moléculaires. C'est la mécanique quantique et le concept de fonction d'onde qui permettent le mieux de comprendre la structure du nuage électronique autour d'un atome. Les électrons d'un atome ne peuvent occuper que certains niveaux d'énergie correspondant à des formes d'ondes déterminées. Ces formes d'onde sont les seules solutions stationnaires de l'équation de Schrödinger autour de cet atome. (Dans le cas de l'atome d'hydrogène, ce sont des harmoniques sphériques.) On donne à ces formes d'onde le nom d'orbitales atomiques (OA).
Au sein d'une molécule, on montre qu'il existe aussi des solutions stationnaires de l'équation de Schrödinger pour les électrons de valence, solutions qui peuvent s'exprimer sous la forme d'une combinaison linéaire des formes d'onde des OA des atomes qui constituent cette molécule. Une telle orbitale est appelée orbitale moléculaire (OM). Une OM peut donner lieu à une liaison chimique si elle permet au doublet d'électrons qui l'occupent d’avoir un niveau d’énergie inférieur.
Dans le cas d'une liaison covalente standard, chaque atome contribue à part égale à l'occupation de l'OM. Dans la molécule dihydrogène H2 par exemple, chaque atome d'hydrogène place un électron dans cette OM. La cohérence de la molécule O2 quant à elle est assurée par deux OM et chaque atome d'oxygène place un électron dans chacune d'entre elles, ce qui fait deux doublets au total.
Dans une liaison entre un cation métallique et un ligand, c'est le ligand qui fournit le doublet qui occupe l'orbitale moléculaire. Le cation se contente de fournir le site de coordination, le puits de potentiel qui conditionne la géométrie de l'orbitale moléculaire. C’est pourquoi on utilise aussi le terme de liaison dative (bien qu’il soit considéré aujourd’hui comme obsolète) : le ligand « donne » une paire d’électrons pour constituer la liaison, le cation est accepteur de la liaison.
Liaison cation métallique-ligand
De nombreux composés complexes se forment autour de cations de métaux de transition. Les métaux de transition sont des éléments du bloc "d" de la classification périodique des éléments. Ils occupent la rangée 4, 5 ou 6 du tableau périodique des éléments et leur bande de valence a une configuration électronique du type (n+1)s2, ndx avec x inférieur à 10 (sous-couche d incomplète). Les éléments de la 7ème rangée ainsi que le lanthane La ne font pas partie de la famille des métaux de transition bien qu'appartenant au bloc d. Le zinc Zn, le cadmium Cd et le mercure Hg ont quant à eux leur sous-couche d remplie et des propriétés chimiques très différentes.
Remarque : certains métaux dits de post-transition (ou métaux pauvres) comme l’aluminium, le gallium, l’étain, le plomb ou l’indium, sont également susceptibles de former des composés complexes.
Les orbitales moléculaires des complexes cations-ligands résultent d'une combinaison linéaire entre une orbitale du ligand et l'une des orbitales (n+1)s, nd et (n+1)p du cation métallique. Leur configuration géométrique dépend du nombre de ligands et du type de recouvrement des orbitales originelles. La configuration la plus stable est obtenue avec 6 ligands en recouvrement sigma de leurs orbitales avec celles du cation métallique. (Un recouvrement sigma est un recouvrement axial. On dit dans ce cas des ligands qu’ils sont sigma-donneurs.) Elle conduit à un octaèdre dont les sommets sont occupés par les ligands, le cation étant au centre. Elle comporte 15 OM auxquelles on a donné les noms suivants :
a1g et a*1g pour les combinaisons avec l’orbitale (n+1)s,
t1u et t*1u (triplement dégénérées) pour les combinaisons avec les orbitales (n+1)p,
eg et e*g (doublement dégénérées) et t2g (triplement dégénérée) pour les combinaisons avec les orbitales nd.
Les orbitales notées avec un astérisque sont anti-liantes (les électrons de cette orbitale se repoussent). Elles ont d'ailleurs une énergie supérieure à celle des orbitales d’origine des ligands. Elles ne donnent pas lieu à une liaison de coordination. Les orbitales t2g ont leur lobe qui n'est pas orienté dans une direction convenable pour assurer un recouvrement sigma. Elles sont non liantes. Il reste donc 6 orbitales liantes possibles pour former un complexe dès lors qu'elles ont une énergie inférieure à celle des orbitales donneuses de doublet des ligands : une orbitale a1g, deux orbitales eg et trois orbitales t1u. Les 3 orbitales non liantes t2g (dont l'énergie n'est pas modifiée) sont occupées en priorité par les électrons appartenant au cation métallique.
Remarque 1 : cette notation fait référence à la théorie des groupes de symétrie octaédrique. Pour qu’il y ait recouvrement entre orbitale du métal et orbitale d’un ligand, il faut qu’elles présentent les mêmes symétries.
Remarque 2 : si l’on s’en tient au raisonnement qui précède, les orbitales a1g, eg et t1u devraient avoir une forme d’onde différente. Une orbitale s n’a pas la même forme qu’une orbitale p. En fait, pour des raisons de symétrie, il y a hybridation de ces orbitales. Ainsi par exemple, au lieu d’avoir une orbitale s et trois orbitales p, on a quatre orbitales sp.
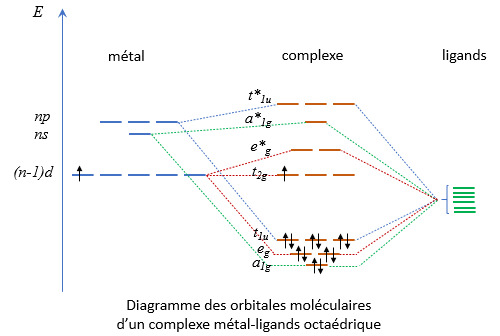
Le titane (III) hexahydraté [Ti(H2O)6]3+, dont le nom officiel est hexaaquo titane (III), illustre ce schéma (voir la figure ci-dessus). L’atome de titane possède 22 électrons (1s2 ; 2s2, 2p6 ; 3s2, 3p6 ; 4s2, 3d2). La configuration électronique du cation Ti3+ est (1s2 ; 2s2, 2p6 ; 3s2, 3p6 ; 3d1). L'électron de valence (3d1) de l'ion Ti3+ reste sur l'une des orbitales t2g alors que les doublets non liants des molécules H2O occupent les orbitales a1g, eg et t1u pour former le complexe [Ti(H2O)6]3+.
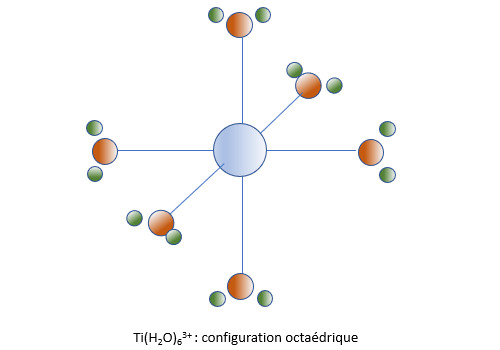
Idem pour l'ion hexacyanoferrate [Fe(CN)6]4-, aussi appelé ferrocyanure. L'ion fer (II) a pour configuration électronique (1s2 ; 2s2, 2p6 ; 3s2, 3p6 ; 4s2, 3d4). Les 6 électrons de sa bande de valence occupent les 3 orbitales t2g du complexe et les doublets non liants des ions cyanures CN- occupent les 6 orbitales liantes a1g, eg et t1u. Le ferrocyanure entre dans la composition du bleu de Prusse, un pigment connu de longue date.
Les ligands
Si l’on se reporte à la présentation de la liaison de coordination ci-dessus, on constatera qu’il y a une grande similarité avec la notion de réaction acide/base de Lewis. Une base de Lewis est une molécule ou un ion capable de donner un doublet d’électron et un acide de Lewis une molécule ou un ion capable de le recevoir. De fait, dans la liste des ligands les plus courants, on retrouvera un grand nombre de bases de Lewis.
Parmi les molécules simples, on peut citer par exemple la molécule H2O et la molécule CO. Dans la molécule H2O, l’atome d’oxygène conserve deux doublets électroniques non liants dans sa bande de valence, ce qui en fait un ligand qui intervient dans de nombreux complexes. La molécule CO quant à elle représente un cas intéressant. La liaison entre carbone et oxygène est triple. Elle est en fait constituée de deux liaisons covalentes standard et d’une liaison de coordination, liaison dans laquelle l’atome d’oxygène joue le rôle de donneur et l’atome de carbone celui d’accepteur. (Ce n’est cependant pas un complexe : il n’y a pas d’atome ou de cation central.) On peut donc considérer que la bande de valence de l’atome de carbone comporte 4 doublets : deux doublets covalents, un doublet de coordination « prêté » par l’atome d’oxygène, et un doublet non liant. Idem pour l’oxygène… qui s’est dépouillé d’un doublet au profit du carbone. La molécule CO comporte donc elle aussi deux doublets non-liants, mais cette fois ils ne sont pas portés par le même atome. L’atome de carbone en porte un et l’atome d’oxygène porte l’autre.
Parmi les ligands inorganiques, on trouve aussi de nombreux ions ou radicaux simples. L’ion cyanure CN- et l’ion hydroxyle OH- en sont des exemples que l’on peut comprendre facilement à partir de ce que nous venons de dire. C’est évident pour l’ion hydroxyle (l’atome d’oxygène possède trois doublets non liants si l’on tient compte de la charge électrique de l’ion). Pour l’ion cyanure, on peut faire un raisonnement par analogie avec la molécule CO. En prenant en compte la charge de l’ion, on a une répartition similaire des doublets entre atome de carbone de carbone et atome d’azote.
Les ions NO2- et NH2- ainsi que l’ammoniac NH3 rentrent également dans la composition de nombreux complexes. C’est chaque fois l’atome d’azote qui est porteur du doublet non liant. Même chose pour les phosphines, phosphinites et phosphonites, avec cette fois l’atome de phosphore qui est porteur du doublet.
Dans ce tour d’horizon des ligands inorganiques, il convient de citer deux cas particuliers importants : celui des ions halogénures et celui de l’ion hydrure. Les halogénures sont connus pour entrer dans la composition de nombreux sels dans lesquels ils entretiennent des liaisons ioniques. Du fait de la constitution de leur bande de valence, les ions halogénures se révèlent également être des ligands forts. La charge électronique qu’ils portent leur permet en effet d’avoir quatre doublets non liants ! Pour ce qui est de l’ion hydrure H-, il est facile de comprendre qu’il engage facilement son doublet d’électrons dans une liaison avec un élément plus électronégatif que lui (ce qui est le cas de tous les cations métalliques).
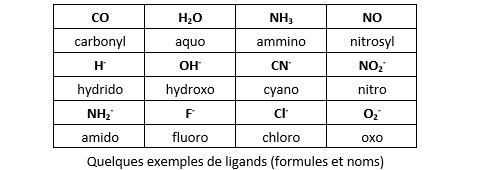
Remarque : on peut classer empiriquement les ligands en fonction de leur énergie de liaison :
S2− < Cl− < F− < O2− < H2O < NH3 < NO− < CN-< CO
Le ligand carbonyle CO est clairement le plus fort et comme on le voit le ligand H2O > a une force moyenne.
Certaines molécules organiques se prêtent à des liaisons de coordination qui peuvent donner lieu à la constitution de complexes. Nous reviendrons sur ce sujet dans le post suivant.
La nomenclature des complexes suit une règle de nommage simple. Lorsqu’il s’agit d’un anion la règle est la suivante :
[nom des ligands] [nom du métal] -ate (degré d’oxydation)
Lorsqu’il s’agit d’un composé neutre ou d’un cation, la règle est :
[nom des ligands] [nom du métal] (degré d’oxydation)
Les ligands sont généralement cités dans l’ordre alphabétique. Exemples :
[Fe(CN)6]4- hexacyanoferrate (II)
[Ti(H2O)6]3+ hexaaquo titane (III)
[PtCl(NH3)5]3+ chloro pentaammino platine (IV)
Géométrie et nombre de coordination
Nous avons jusqu’à présent cité en exemple des complexes comportant 6 ligands. C’est la configuration la plus répandue mais il en existe d’autres en fonction du nombre de ligands :
linéaire avec 2 ligands comme le diamine cuivre [Cu(NH3)2]+,
tétraédrique avec 4 ligands comme le tétracarbonyle de nickel Ni(CO)4,
plane carrée avec 4 ligands comme le tétracyanure de nickel [Ni(CN)4]2-,
bipyramide trigonale avec 5 ligands comme l’hydrocarbonyle de cobalt hydrocarbonyle de cobalt HCo(CO)4 ,
etc...
On appelle nombre de coordination le nombre d’atomes auquel le cation central est lié.

Règle des 18 électrons
La règle des 18 électrons stipule que les complexes métalliques les plus stables sont obtenus lorsque la couche de valence des cations au centre du complexe est saturée. Pour un métal de transition, cette couche peut accueillir 18 électrons, d’où le nom de cette règle.
Cette règle des 18 électrons est loin de s’appliquer de manière systématique. Il semble que la stabilité des complexes soit plutôt liée à des considérations de symétrie, les formes géométriques comme celles indiquées ci-dessus étant privilégiées, que la règle soit respectée ou pas. Quelques exemples vont nous permettre d’illustrer notre propos.
Configuration tétraèdrique
Le tétracarbonyle de nickel Ni(CO)4 est l’exemple typique de complexe tétraédrique dont la bande de valence est remplie. Le tétrachlorure de titane (IV) TiCl4, par contre, est loin de faire le plein. Le cation central Ti4+ dont la configuration électronique est (4s0, 3d0) n’accueille que 4 paires d’électrons, soient 8 électrons au total sur les 18 possibles dans sa bande de valence. Le tétrachlorure de vanadium (IV) VCl4 présente lui aussi un fort déficit en électrons de la bande de valence du vanadium (9 électrons au lieu de 18). Même chose pour le dichlorure de titanocène (C2H5)2TiCl2. Le titanocène est un métallocène, un complexe dans lequel le cation est relié à deux ligands cyclopentadiényles par une liaison éta (les anions C5H5 sont aromatiques).
Nota : les composés complexes dont les ligands sont des ions chlorure ou fluorure n’ont le plus souvent pas besoin de la règle des 18 pour être stables.
Plan carré
Dans le tétracyanure de nickel (II) [Ni(CN)4]2- la configuration de l’ion Ni2+ est (3d8). Les ions cyanure qui forment un carré autour de lui ne lui apportent que 3 autres paires d’électrons et le compte n’y est pas... Dans le cis-diammino dichloro platine (II) Pt Cl2(NH3)2 le platine, qui est oxydé deux fois, conserve également une orbitale libre. Le cis-diammino dichloro platine est aussi appelé CDDP ou cisplatine. C’est un médicament anticancéreux.
Nota : le préfixe cis- indique que les ligands chloro sont adjacents. Dans le cas contraire, on utilise le préfixe trans-.
Bipyramide trigonale
L’hydrocarbonyle de cobalt HCo(CO)4 a une configuration bipyramide trigonale. Le cobalt a la configuration électronique (4s2, 3d7). Il fait le plein avec les quatre doublets apportés par les groupes CO et l’électron célibataire de l’atome d’hydrogène. Idem pour l’octacarbonyle de dicobalt Co2(CO)8 dans lequel il se décompose à l’ambiante et qui forme un empilage de deux bipyramides trigonales.
Octaèdre
Les composés octaédriques qui satisfont à la règle des 18 sont nombreux. L’hexaaquo fer (II) [Fe(H2O)6]2+ la respecte mais pas l’hexaaquo titane [Ti(H2O)6]3+. Dans le composé HMn(CO)5 le manganèse fait le plein, de même que dans le composé le décacarbonyle de dimanganèse Mn2(CO)10 composé d’un empilage de deux octaèdres.
Dans l’hexa-amine chrome (III) [Cr(NH3)6]3+ le chrome conserve trois électrons célibataires (15 électrons au lieu de 18). Dans l’hexacarbonyle de vanadium V(CO6), le vanadium (4s2, 3d3) accueille quant à lui 6 doublets non liants, soient 17 électrons au lieu de 18.
Le chlorure de molybdène (V) (MoCl5)2 est constitué de deux octaèdres qui partagent deux ions chlorure de leur base carré. La configuration électronique du cation molybdène (V) est (5s1). Dans le chlorure de molybdène, sa bande de valence n’accueille donc que 4 doublets et deux électrons non appariés supplémentaires. Il ne fait pas le plein.

Dans le réactif de Schweizer [Cu(NH3)4(H2O)2]2+ l’ion Cu2+ possède 9 électrons dans sa bande de valence. Il se trouve donc largement en excès par rapport à la règle des 18 électrons ! L’explication réside dans le fait que les liaisons autour du cuivre ont une forte composante ionique.
Le composé hexa-aquo fer (III) [Fe(H2O)6]3+ est un cas tout à fait particulier. Son caractère nettement paramagnétique incite à penser qu’il conserve les 5 électrons célibataires de la sous-couche 3d de l’ion fer (III) (voir plus bas les propriétés magnétiques des complexes). Ceci ne peut s’expliquer que si deux orbitales 4d sont mobilisées par le composé. L’octaèdre serait alors constitué par l’hybridation de l’orbitale 4s, des trois orbitales 4p et de ces deux orbitales 4d (hybridation sp3d2).
Complexes multi-métalliques
Nous avons jusqu’à présent supposé que le corps central était un atome ou un cation métallique. Les atomes métalliques se combinent facilement entre eux et il existe des complexes polyatomiques dont le noyau central est un polymère Mn (M étant un atome métallique). C’est le cas par exemple du dodécarbonyle trifer Fe3(CO)12. Le noyau central est constitué par trois atomes de fer formant un triangle. Chaque atome est porteur de quatre liaisons de coordination avec des ligands carbonyles CO. La configuration du fer reste du type octaédrique (6 liaisons). Dans le dodécarbonyle tétracobalt Co4(CO)12, les quatre atomes de cobalt occupent le sommet d’un tétraèdre et portent chacun trois liaisons de coordination avec des ligands carbonyles.
Couleur et propriétés magnétiques
Les composés complexes ont une couleur franche (certains sont utilisés comme pigment) et des propriétés magnétiques bien définies. Les différents niveaux d’énergie des orbitales permettent d’expliquer ces propriétés (on utilise dans ce cas la théorie des champs cristallins qui est une adaptation de la théorie des OA au cas d’un réseau). La couleur d’un complexe est la couleur complémentaire de la fréquence absorbée lors d’un saut d’un niveau à un autre. Les propriétés magnétiques quant à elles s’expliquent par la différence d’énergie entre les différentes orbitales.
Les électrons d’une même couche ont tendance à se répartir sur les différentes orbitales de façon à minimiser le nombre d’électrons appariés (règle de Hund). Cela résulte du fait que l’appariement des électrons (c.à.d le fait deux électrons de spin opposé occupent la même orbitale) a un coût énergétique. Dans la figure représentant l’énergie des orbitales moléculaires nous avons supposé qu’il y avait un appariement maximal (règle de Hund non respectée). Ceci dépend en fait de l’écart d’énergie entre les orbitales. Si le coût énergétique de l’appariement des électrons est supérieur à cet écart, les électrons vont avoir tendance à se répartir entre les deux niveaux pour minimiser leur énergie totale. La règle de Hund sera alors respectée. Dans le cas contraire (comme dans la figure) ils vont s’apparier. On caractérise ces deux situations en disant que la première est de type champ faible – spin fort et la seconde champ fort - spin faible. Cela a une incidence directe sur les propriétés magnétiques. Un complexe champ faible – spin fort est paramagnétique alors qu’un complexe champ fort – spin faible est diamagnétique. Question de spin ! On peut d’ailleurs remarquer qu’un complexe champ fort – spin faible est souvent incolore. Le delta d’énergie pour faire passer un électron sur un niveau orbitalaire d’énergie supérieure est souvent trop élevé pour être dans le domaine visible.
Pour en savoir plus :
post d’introduction à la chimie
post sur les électrons
post sur les électrons dans les liaisons covalentes
post sur les liaisons chimiques
post sur les orbitales moléculaires
post sur la valence
post sur la géométrie des molécules
post sur la cohésion de la matière
post sur la classification périodique des éléments
post sur les métaux de transition
post sur les acides et bases
post sur les complexes et les ligands : l’approche covalente
post sur les complexes et les ligands : exemples
post sur le phosphore
post sur l’azote
post sur les matériaux magnétiques
index
2 notes
·
View notes
Text
Métaux alcalins et alcalino-terreux
Métaux alcalins
On donne le nom de métaux alcalins aux éléments de la première colonne du tableau périodique des éléments en dehors de l’hydrogène. Ces éléments sont le lithium Li, le sodium Na, le potassium K, le rubidium Rb, le césium Cs et le Francium Fr. Ils ont tous la particularité de n’avoir qu’un seul électron dans leur bande de valence. De fait, ils sont tous très peu électronégatifs, beaucoup moins que l’hydrogène par exemple dont le potentiel d’électronégativité est égal à 2,2. Ils sont nucléophiles (donneurs d’électron). Le francium est un élément radioactif de durée de vie égale à 21 minutes.
Le sodium, le potassium et le fluor sont des oligoéléments. Is sont respectivement en 9è, 7è et 13è position dans la liste des éléments les plus abondants du corps humain. Dans la croûte terrestre, le sodium et le potassium pointent en 6è et 7è place (28000 et 26000 ppm). En revanche, ils sont peu abondants l’univers, le lithium ne représentant que 2,3 10-6 de la masse baryonique et le sodium 1 10-6.
Dans les conditions normales de température et de pression, si l’on excepte le francium qui fond à 20°, ils se présentent sous la forme d’un métal plutôt mou (on peut le couper avec un couteau). Ils s’oxydent très vite à l’air libre, perdant ainsi l’aspect brillant que leur confère leur caractère métallique. Ils sont relativement léger : la densité des trois premiers (Li, Na, K) est inférieure à 1, celle des trois autres inférieure à deux. Leur point de fusion s’étage entre 20° pour le francium et 97,8° pour le sodium. Seul le lithium reste solide jusqu’à 180°. Leur point d’ébullition est supérieur à 650°, celui du lithium atteint 1342°.
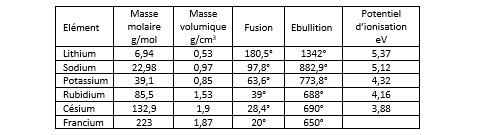
Le potentiel de première ionisation des métaux alcalins est relativement faible : de 3,88 eV pour le césium à 5,37 eV pour le lithium. Ils s’ionisent donc facilement pour donner les cations Li+, Na+, K+, Rb+ et Cs+. Ceci explique leur caractère nucléophile. Il est beaucoup plus difficile de leur arracher un deuxième électron (27 eV pour le césium, 47 eV pour le sodium). Il faut en effet aller chercher cet électron sur la couche n-1 qui est complète, donc très stable.
Le potentiel d’oxydoréduction du lithium est élevé, ce qui en fait un composant très recherché pour réaliser des piles (piles au lithium).
Hydroxyde
Les métaux alcaline réagissent violemment avec l’eau en dégageant de l’hydrogène qui brûle dans l’air et peut même provoquer une explosion :

L’hydroxyde de sodium NaOH est plus connu sous le nom de soude caustique. La soude caustique se présente sous forme de cristaux. Elle est très hygroscopique. En solution, elle libère l’ion OH- qui est une base forte. Elle est utilisée de manière intensive, en particulier pour ses propriétés de détergent. L’hydroxyde de potassium KOH porte le nom de potasse. Il est utilisé en industrie chimique pour produire des engrais ou du savon.
La lithine LiOH est moins connue, c’est une base forte très corrosive. En présence de peroxyde d’hydrogène, l’hydroxyde de lithium forme un composé ionique appelé hydroperoxyde de lithium LiOOH :
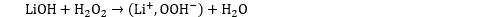
Hydrures
Les métaux alcalins réagissent avec l’hydrogène pour former des hydrures :

Les hydrures de métaux alcalins sont des bases fortes qui libèrent des ions OH- en solution dans l’eau.
Oxydes
Les métaux alcalins s’oxydent rapidement pour donner des oxydes, des peroxydes ou des superoxydes :
Li2O, Li2O2
Na2O, Na2O2, NaO2
K2O, K2O2, KO2
Cs2O, Cs4O
Ces oxydes sont en général basiques et réagissent avec l’eau pour donner des hydroxydes. Les peroxydes et superoxydes ont tendance à redonner naturellement des oxydes et de l’oxygène.
La bande de valence du soufre a la même configuration que celle de l’oxygène, il réagit avec les métaux alcalins pour donner des sulfures (sulfure de sodium Na2S, sulfure de potassium K2S). Ces sulfures s’hydrolysent complètement pour donner des hydroxydes :

L’ion SH- a des propriétés très proches de celles de l’ion OH-.
Halogénures
Nucléophiles, les métaux alcalins sont attaqués par les halogènes pour former des halogénures. Le chlorure de sodium NaCl et le chlorure d potassium KCl sont bien sûr connus pour leur utilisation culinaire. Le chlorure de lithium LiCl est utilisé dans l’industrie chimique et sidérurgique. Les fluorures (LiF, NaF, KF) sont toxiques. Ils sont utilisés dans l’industrie. L’iodure de potassium KI a de nombreuses applications en chimie organique, dans l’alimentation mais aussi dans l’industrie pharmaceutique.
Sels d’oxacides
Du fait de leur caractère nucléophile les métaux alcalins forment des sels avec tous les oxacides. Des nitrates et des nitrites tout d’abord. Si le nitrate de sodium NaNO3 et le nitrate de lithium LiNO3 ont un emploi limité, le nitrate de potassium KNO3, plus connu sous le nom de salpêtre, a longtemps été utilisé comme ingrédient principal pour faire de la poudre à canon. Il continue d’être utilisé comme propergol ou en charcuterie. Le nitrite de sodium NaNO2 a lui aussi des applications alimentaires ainsi que dans l’industrie chimique.
Le carbonate de potassium K2CO3 est utilisé comme engrais et pour ses propriétés fongicides. Il rentre également dans la composition du verre. Le carbonate de sodium Na2CO3 (appelé improprement cristaux de soude) a de nombreuses applications (savon, sidérurgie…). Le bicarbonate de soude (Na+CO2OH-) et le bicarbonate de potassium (K+CO2OH-) ont des usages multiples, y compris domestiques comme régulateur de pH. Ce sont des hydrogénocarbonates. On les prépare en faisant réagir le carbonate correspondant avec de l’eau et du dioxyde de carbone :

Le carbonate de lithium Li2CO3 a quelques applications industrielles.
Le sulfate de sodium Na2SO4 est produit industriellement pour l’industrie du verre ou celle des textiles. Ses propriétés de détergent sont également appréciées. Le sulfate de potassium K2SO4, que l’on trouve dans les plantes, est un engrais. Il est aussi utilisé comme composant dans les peintures. Le sulfite de sodium Na2SO3 est un additif alimentaire aux vertus antioxydantes. Le thiosulfate Na2S2O3 est un ligand. Il est utilisé comme fixateur en photographie mais également comme antidote pour neutraliser l’effet de certains produits toxiques.
Le phosphate de sodium Na3PO4 est un additif alimentaire, tout comme le dihydrogénophosphate de potassium KH2PO4. L’hydrogénophosphate de potassium K2HPO4 est utilisé comme régulateur d’acidité de l’estomac et dans l’agriculture biologique.
On ne peut pas terminer ce tour d’horizon des sels alcalins sans parler du permanganate de potassium KMnO4. Connu pour ses vertus de désinfectant, il est aussi utilisé dans la conservation des fruits (il arrête le processus de mûrissement en oxydant l’éthylène dégagé par les fruits) et le traitement de eaux. C’est aussi un composant intervenant dans la fabrication de certains ergols. permanganate de calcium CaMnO4. C’est un agent blanchissant que l’on trouve dans la composition de nombreux dentifrices.
Métaux alcalino-terreux
Les métaux alcalino-terreux sont les éléments de la deuxième colonne. Ces éléments sont le béryllium Be, le magnésium Mg, le calcium Ca, le strontium Sr, le baryum Ba et le radium Ra. Le radium est radioactif. La demi-vie du 226Ra est de 1600 ans. Leur caractéristique commune est de posséder deux électrons dans leur bande de valence.
Le magnésium est relativement abondant dans l’Univers (0,06%). Il se classe à la 9ème position à l’échelle de la voie lactée. La calcium figure dans la liste des 20 premiers (0,007%). Les autres éléments sont beaucoup plus rares. Le magnésium et le calcium sont les seuls métaux alcalino-terreux relativement abondants dans la croûte terrestre : 2,9% pour le magnésium et 5% pour le calcium. Le calcium est un outre un oligoélément qui représente 1,4% de la masse du corps humain (en particulier dans le squelette).
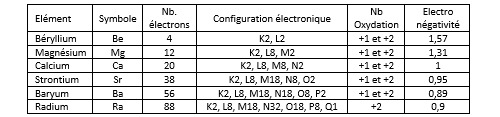
Dans les conditions normales de température et de pression ils se présentent sous forme cristalline. Si l’on excepte le béryllium, le point de fusion des métaux alcalino-terreux est inférieur à 1000°C. Le béryllium, le magnésium et le calcium (et même le strontium) sont peu denses.
Comme les métaux alcalins ils sont peu électronégatifs. Si l’on excepte le béryllium qui noue généralement des liaisons covalentes, Ils donnent facilement des cations chargés deux fois positivement : Mg2+, Ca2+...
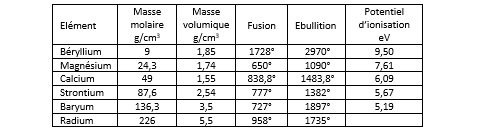
Oxydes
Les oxydes des métaux alcalino-terreux (BeO, MgO, CaO, SrO, BaO) sont des oxydes cristallins de couleur blanche. Ils sont obtenus par combustion du métal dans le dioxygène, par calcination d’un sel, carbonate ou nitrate, ou par déshydratation d’un hydroxyde. Leur température de fusion est très élevée (supérieure à 2500°C pour l’es oxydes de béryllium, de magnésium et de calcium). L’oxyde de calcium est appelé communément chaux vive.
Halogénures
Les métaux alcalino-terreux sont très réactifs avec les halogènes. Si l’on excepte le béryllium, ils forment des sels ioniques qui s’hydrolysent dans l’eau. Les halogénures de béryllium (comme le chlorure de béryllium BeCl2) sont quant à eux covalents et ne réagissent pas avec l’eau.
Hydroxydes
Le magnésium, le calcium, le strontium et le baryum forment des hydroxydes qui sont des bases fortes :

L’hydroxyde de calcium est appelé chaux éteinte. Le béryllium ne réagit pas avec l’eau.
Sels d’oxacides
Magnésium, calcium, strontium et baryum forment aisément des sels d’oxacides (carbonate, nitrate, phosphate, sulfate) et c’est en général sous cette forme qu’on les trouve dans la nature. Le carbonate de calcium CaCO3 par exemple est le composant de base du calcaire et le gypse est un sulfate de calcium hydraté CaSO4.2H2O. Le béryllium fait une nouvelle fois exception du fait de sa faible propension à former de tels composés.
Composés organomagnésiens
Les composés organomagnésiens sont des molécules organiques dans laquelle il y a au moins une liaison carbone-magnésium. Parmi les composés organomagnésiens on distingue une sous-famille appelée réactifs de Grignard de formule générique R-MgX, R étant une molécule organique et X un halogène.
Les composés organomagnésiens jouent un rôle important en chimie organique et notamment en synthèse organique. Les réactifs de Grignard permettent en particulier de réaliser des substitutions nucléophiles et des additions nucléophiles.
Applications
La magnésium est utilisé en mécanique sous forme d’alliage avec l’aluminium (le magnésium est inflammable et est rarement utilisé seul). Il est aussi utilisé en chimie organique (voir ci-dessus). Le calcium est utilisé pour ses propriétés réductrices dans le traitement du minerai ainsi que dans la production du plâtre et du ciment. En tant qu’oligoéléments, le magnésium et le calcium sont utilisés comme composants dans des produits pharmaceutiques et parapharmaceutiques. Le béryllium a des applications militaires (rigidité et stabilité sur une large gamme de température). L’oxyde de béryllium qui est à la fois isolant et conducteur thermique a des applications en microélectronique. Les autres métaux alcalino-terreux ont beaucoup moins d’applications.
Rôle dans la chimie du vivant
Le calcium et le magnésium jouent un rôle essentiel dans la chimie du vivant. L’hydroxyapatite Ca5(PO4)3(OH)2 est le composant minéral qui donne leur solidité aux os et aux dents. Le carbonate de calcium forme la coquille des mollusques. Calcium et magnésium sont aussi des oligoéléments. Le magnésium en particulier est un cofacteur de nombreuses enzymes.
Pour en savoir plus :
post sur le tableau périodique des éléments
post sur l’électronégativité
post sur les espèces nucléophiles et électrophiles
post sur les acides et les bases
post sur les sels
post sur les halogènes
post sur le calcium
post sur le mégnésium
index
#chimie#minéraux#alcalin#sodium#potassium#lithium#hydrure#hydroxyde#acide#électronégativité#nucléophile#rubidium#césium#sel#nitrate#phosphate#oxyde#oxydoréduction
0 notes
Text
Oxydoréduction
Une oxydoréduction est une réaction au cours de laquelle il se produit un transfert d’électrons. On dit de l’élément (ou du composé) qui donne un ou plusieurs électrons qu’il est oxydé et de l’élément ou du composé qui accepte ce(s) électron(s) qu’il est réduit. Un élément ou un composé qui a une forte propension à accepter (voire à chiper) des électrons est un oxydant. L’élément ou le composé donneur est appelé réducteur. Ces deux éléments forment un couple redox.
Le rôle des électrons dans les liaisons chimiques
Il existe différents types de liaisons chimiques (voir les posts à ce sujet). Nous allons nous intéresser à deux d’entre eux : la liaison covalente et la liaison ionique. Dans la liaison covalente, chaque élément met en commun un électron. L’orbitale des deux électrons mis en commun est étendue à la molécule tout entière, ce qui assure la stabilité de la liaison. Dans la liaison ionique, l’un des éléments du composé chimique capte l’un des électrons de l’autre élément et la liaison qui se forme est de nature électrostatique entre un cation et un anion. La différence entre ces deux types de liaison n’est cependant pas aussi tranchée qu’il y paraît. Dans le cas de la liaison covalente, l’un des éléments va souvent « tirer la couverture à lui ». En clair, cela signifie que l’orbitale électronique des électrons engagés dans la liaison ne sera pas répartie de manière symétrique entre les deux éléments. La probabilité de trouver ces électrons est plus forte près de l’un des deux éléments que de l’autre. Dans ce cas, la liaison covalente est donc partiellement ionique. C’est le cas de l’eau (H20) mais également, par exemple, du dioxyde de carbone (CO2) ou de l’ammoniac (NH3).
Oxydation et réduction
Lorsqu’Antoine Lavoisier établit au XVIIIème les premières définitions des réactions d’oxydation et de réduction, les notions d’atome, d’électron et de liaison covalente n’étaient pas connues. Pour Lavoisier, l’oxydation était la combinaison d’un élément avec l’oxygène et la réduction l’extraction d’un élément à partir d’un oxyde :

On a remarqué plus tard la similitude de certaines réactions avec les réactions d’oxydation et de réduction telles que les avait définies Lavoisier. Ainsi par exemple :

La tentation était grande de dire que le chlore oxyde le cuivre. Tout comme on peut dire que l’hydrogène réduit l’azote dans la réaction qui produit l’ammoniac.
Potentiel d’oxydoréduction
La nature « électrique » des phénomènes intervenant dans les réactions d’oxydoréduction est clairement apparue lorsque les premières piles électriques ont été mises au point : d’abord en 1800 par Alessandro Volta puis en 1836 par John Daniell. La pile Daniell est la première avoir été utilisée de manière industrielle. Elle comporte deux compartiments. Chaque compartiment contient un couple redox. Un coupe redox est constitué d’un oxydant et de sa forme réduite. Dans le cas de la pile Daniell, le compartiment relié au pôle négatif (anode) contient une lame de zinc dans une solution de sulfate de zinc et le compartiment relié au pôle positif (cathode) une lame de cuivre dans une solution de sulfate de cuivre. Les deux compartiments sont reliés chimiquement par un pont salin contenant une solution de chlorure de potassium KCl ou de nitrate de potassium KNO3. Lorsque les deux pôles sont reliés à un circuit électrique extérieur, il apparaît une différence de potentiel entre le pôle plus (la cathode) et le pôle moins (l’anode) qui permet la circulation d’un courant. Le processus générateur de courant peut être décomposé sous la forme de deux demi-réactions :

Le courant qui circule s’accompagne d’une réaction d’oxydation du zinc et de réduction du sulfate de cuivre : il y a croissance de la lame de cuivre et dégradation de la lame de zinc (voir le post sur les piles alcalines).
Il faudra attendre le début du XXème siècle pour que l’on comprenne de façon plus précise le rôle des électrons dans les réactions d’oxydoréduction mais on n’a pas attendu cette date pour établir une échelle de potentiel d’oxydoréduction. Le potentiel d’oxydoréduction est une grandeur empirique, exprimée en Volt, obtenue en mesurant le potentiel électrique d’une pile composée du couple redox dont on veut mesurer le potentiel et d’un couple redox de référence, en l’occurrence le couple H+/H2. Ce couple est aussi appelé électrode normale à hydrogène (ENH). Lorsque le courant circule depuis l’électrode plongée dans le couple à mesurer vers l’ENH le potentiel est positif et il est négatif dans le cas contraire. Autrement dit, lorsque ce potentiel, noté E0, est positif, le couple a un caractère plutôt oxydant et lorsqu’il est négatif, il a un caractère plutôt réducteur.
Réaction d’oxydoréduction et couple redox
Sur cette base, on peut dire qu’une réaction d’oxydoréduction est une réaction qui fait intervenir deux couples redox (Oxy1, Red1) et (Oxy2, Red2) :

Dans cette réaction, le transfert d’électrons se fait de l’élément Red2 vers l’élément Oxy1. Comme on l’a fait dans le cas de la pile Daniell, on peut décomposer cette réaction en deux demi-réactions :

En termes de potentiels d’oxydoréduction, cette réaction est rendue possible parce qu’il existe une différence de potentiel redox favorable :

Si on reprend l’exemple de la pile Daniell, le potentiel du couple (Zn++/Zn) est négatif et il vaut -0,76 V. Le potentiel du couple (Cu++/Cu) est quant à lui positif et il vaut 0,34 V. Si on verse une solution de sulfate de cuivre dans un tube à essai contenant la poudre de zinc, la solution initialement bleutée devint incolore et il apparait une poudre orangée caractéristique de la présence de poudre de cuivre :

Le tube chauffe de manière significative (la réaction est exothermique). Une différence de potentiel redox positive entraîne, la plupart du temps, une réaction spontanée d’oxydoréduction. Prenons l’exemple de l’oxydation du fer par l’oxygène :

Les couples redox intervenant dans cette réaction sont :
(O2/O2-) dont le potentiel redox est 1,12 V,
(Fe3+/Fe) dont le potentiel redox est -0,04 V.
On vérifie bien que la différence de potentiel redox est positive, ce qui correspond à une oxydation spontanée du fer par le dioxygène. Nous avons dit en début de ce post que le chlore se comportait la plupart du temps comme un oxydant. Le potentiel redox du couple (Cl2/Cl-) est en effet de 1,36 V, il est donc a priori encore plus oxydant que l’oxygène :
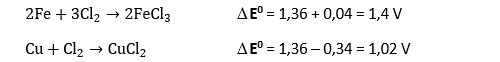
En toute rigueur, le terme E0 doit être corrigé en fonction de différents paramètres qui tiennent compte de l’environnement et de la concentration en éléments chimiques en utilisant une formule appelée équation de Nernst :

R étant la constante des gaz parfaits, F la constante de Faraday et les termes aox et ared pouvant être approximés par les valeurs de concentration des éléments considérés. L’équation de Nernst permet en particulier d’établir les conditions dans lesquelles il y a équilibre entre les deux membres de la réaction. Walther Nernst est un physicien et chimiste allemand qui vécut entre 1864 et 1941.
Couples redox
Les chimistes ont établi des listes de couples redox avec leur potentiel d’oxydoréduction. Il en existe une très grande variété et nous n’en donnerons ici que quelques exemples :

Et cetera… A chacun de ces couples, nous l’avons dit, est associé un potentiel soigneusement consigné dans ces listes. On trouvera ci-dessous une liste non exhaustive de potentiels standards :

Pour en savoir plus :
post d’introduction à la chimie
post sur les éléments chimiques
post sur les liaisons chimiques
post sur le degré d’oxydation
post sur le haut-fourneau
post sur la cohésion de la matière
post sur les piles alcalines
post sur les acides et les base
glossaire de chimie générale
post sur la classification périodique des éléments
index
#oxydation#réduction#Oxydoréduction#redox#chimie#oxygène#hydrogène#pile alcaline#daniell#volta#lavoisier#potentiel#cuivre#zinc#oxyde#hydroxyde#acide#sel#hydracide#oxacide#nernst
0 notes
Text
Acides et bases
La définition la plus commune des acides a été proposée en 1923 par Nicolaus Brönsted. Pour Brönsted, un acide est un composé chimique capable de libérer un proton H+ pour donner une base. La définition de Bronsted reprend et généralise une définition plus restrictive proposée à la fin du XIXème siècle par Svante Arrhénius. Dans son acception la plus courante, un couple acide-base est noté AH/A- :

Remarque : le proton H+ se rencontre rarement seul. Il est le plus souvent solvaté, par exemple sous la forme d’un ion hydronium H3O+.
On dit de A- que c’est la base conjuguée de l’acide AH. L’acide chlorhydrique HCl et l’acide sulfurique H2SO4 en sont les exemples les plus emblématiques. L’acide chlorhydrique est un composé gazeux soluble. Sa base conjuguée est l’ion Cl-. L’acide sulfurique existe lui sous forme liquide. Sa base conjuguée est l’ion Sulfate SO42-. Dans le cas de ces deux acides, la libération de proton se fait par dissociation. Elle peut également se faire par décomposition dans l’eau. On note ces couples sous la forme B+/BOH :

La notion de base est, d’une certaine manière, définie en miroir de celle d’acide. Une base est une espèce capable de capter un proton pour donner un acide. On rencontre souvent dans la littérature une définition différente de cette notion. Elle alors présentée comme une substance chimique capable de libérer des ions hydroxyde OH- :

Cette définition qui renvoie à la formulation d’Arrhénius est plus restrictive puisqu’elle ne s’applique qu’en solution aqueuse. Elle s’applique bien à certains composés comme les hydroxydes. L’hydroxyde de sodium NaOH par exemple, un solide blanc plus connu sous le nom de soude, est une base dont la forme acide conjuguée est l’ion Na+. Elle ne s’applique pas à l’ammoniac NH3 qui est tout à fait capable de capturer un proton en dehors de tout contexte humide :

La molécule NH3, qui est polaire, attire le proton faiblement lié au chlore dans le chlorure d’hydrogène pour former un ion NH4+ qui attire à son tour l’ion Cl- (ammoniac et chlorure d’hydrogène sont à l’état gazeux). Il se forme à l’issue de la réaction un brouillard blanchâtre constitué de fines particules de chlorure d’ammonium. L’ammoniac est donc bien une base au sens de Bronsted tout en échappant à la formulation d’Arrhénius.
L’eau a un statut particulier. L’eau peut aussi bien jouer le rôle d’un acide que d’une base. On dit que c’est un ampholyte (l’adjectif associé est amphotère). L’eau est capable de perdre un proton pour donner l’ion hydroxyde HO¯ et peut aussi gagner un proton pour former l'ion hydronium H3O+. L’eau est donc un acide dans le couple H2O/HO¯ et une base dans le couple H3O+/H2O. Acide et base de Lewis
Le chimiste Gilbert Lewis a donné une définition différente de la notion d’acide et de base. Une base de Lewis est un composé chimique donneur d’un doublet d’électrons et qui possède donc un tel doublet électronique non liant libre dans sa bande de valence. Un acide de Lewis est un composé chimique accepteur de doublet électronique. Un acide de Lewis est donc un composé chimique qui présente structurellement une orbitale vide.
Prenons le cas du borane BH3. Le bore possède 5 électrons : deux sur sa couche n = 1 et 3 sur sa couche n = 2 (répartis sur les sous-couches l = 0 et 1). Ces trois électrons sont engagés dans les liaisons avec les atomes d’hydrogène et les trois doublets ainsi constitués occupent trois des orbitales de la couche n = 2. Or il y a 4 orbitales possibles sur cette couche. Il en résulte que le borane dispose d’une orbitale libre et peut accepter un doublet supplémentaire. C’est donc un acide de Lewis. Même cas de figure pour le chlorure d’aluminium AlCl3 ou le fluorure d’aluminium AlF3. L’aluminium possède 13 électrons, 2 sur sa couche n = 1 et 8 sur sa couche n = 2, qui sont toutes deux saturées. Il reste 3 électrons sur sa couche de valence, répartis sur les sous-couches l = 0 et l = 1. Ces trois électrons sont engagés dans les liaisons avec les atomes de chlore ou de fluore, ce qui fait trois doublets occupant trois des orbitales des sous-couches l = 0 et l = 1 qui peuvent en comporter 4. Le chlorure d’aluminium et le fluorure d’aluminium sont également des acides de Lewis.
L’azote de son côté possède 7 électrons, deux sur sa couche n = 1 et 5 sur sa couche n = 2. Dans la molécule d’ammoniac NH3, 3 de ces électrons sont engagés dans les liaisons avec les atomes d’hydrogène, formant ainsi ce que l’on appelle des doublets liants. Les deux derniers électrons constituent un doublet non liant qui fait de l’ammoniac une base de Lewis. En présence d’un catalyseur, l’ammoniac réagit avec la borane pour donner du borazane :

La définition de Lewis recouvre celle de Bronsted : un proton H+ n’est-il pas l’acide de Lewis le plus élémentaire ? L’inverse n’est pas nécessairement vrai.
Nota : le test du pH (voir plus bas) ne s’applique pas aux acides de Lewis. Cependant, l'introduction d'un acide de Lewis dans l'eau conduit la plupart du temps à une diminution de son pH.
Acide fort, acide faible et pH
Nous avons souligné plus haut le fait que les acides se dissociaient dans les solvants, transformant le solvant en un acide :

On dit d’un acide qu’il est fort lorsqu’une telle réaction est complète et rapide. Dans ce cas le composé AH n’existe pas en solution car il est entièrement dissocié. Lorsque la réaction conduit à un équilibre entre les deux membres de la réaction, on a affaire à un acide faible. On définit de la même façon la notion de base forte ou faible en fonction de la rapidité et de la complétude de la dissociation de celle-ci dans un solvant.
Cette notion de force des acides et des bases se mesure à l’échelle du pH (potentiel hydrogène) de la solution :

aH étant l’activité des ions hydrogène H+. L’activité est assimilable, en première approximation, à la concentration en ion hydronium H3O+. Le pH varie entre 0 et 14. Un acide fort très peu dilué à un pH faible, voire égal à zéro. Une base forte très peu diluée a un pH voisin ou égal à 14. Une solution de pH égal à 7 est neutre.
Différents types d’acide
En chimie minérale, on distingue deux classes d’acides : les hydracides et les oxacides. Les hydracides (qu’on appelle aussi acides binaires) sont composés d’un non-métal associé à un ou plusieurs atomes d’hydrogène. Leur formule générique est HX. Il existe deux façons de les nommer. Soit <X-nom> le nom du non-métal considéré :
On peut les nommer en faisant suivre <X-nom> par le suffixe -ure et en ajoutant la mention « d’hydrogène ». Exemples : chlorure d’hydrogène HCl, sulfure d’hydrogène H2S.
On peut aussi les nommer en formant un adjectif avec <X-nom> suivi du suffixe -hydrique. On utilise cette dénomination de préférence lorsque l’acide considéré est en solution aqueuse. Exemples : acide chlorhydrique HCl, acide sulfhydrique H2S.
L’acide cyanhydrique HCN (cyanure d’hydrogène) n’est pas, stricto sensu, un acide binaire mais il en a toutes les caractéristiques : le carbone et l’azote sont tous deux des non-métaux et ils sont liés par une triple liaison covalente très forte.
L’anion libéré par un hydracide en solution porte le nom du non-métal suivi du suffixe -ure : ion chlorure Cl-, ion sulfure S2-, ion cyanure CN-.
Les oxacides (ou oxoacides) ont pour formule générique HXO. L’élément noté X est le plus souvent un non-métal mais il existe des oxacides à base de métal fortement oxydé (acide manganique HMnO4 par exemple). L’élément associé à l’hydrogène dans un oxacide peut être oxydé à différents degrés. On connaît par exemple plusieurs types d’oxacides sulfuriques : l’acide sulfureux H2SO3, l’acide sulfurique H2SO4 et l’acide peroxydisulfurique H2S2O8. La règle doit donc tenir compte du nombre d’oxydation de l’élément oxydé. Le nombre d’oxydation est le nombre de charges électriques (réelles dans le cas d’une liaison ionique, fictive dans le cas d’une liaison covalente) portées par l’élément à la base de l’oxacide. Dans le cas général, le nombre d’oxydation est égal à 1 ou 2. La règle est alors :
de les nommer en faisant suivre <X-nom> par le suffixe -ate ou -ite et en ajoutant la mention « d’hydrogène ». Exemples : nitrite d’hydrogène HNO2, nitrate d’hydrogène HNO3 ;
de les nommer en formant un adjectif avec <X-nom> suivi du suffixe -ique ou -eux. On utilise cette dénomination de préférence lorsque l’acide considéré est en solution aqueuse. Exemples : acide nitreux HNO2, acide nitrique HNO3.
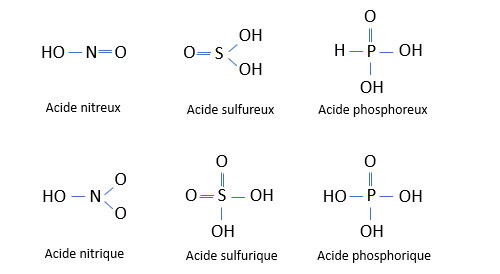
Comme on le voit dans la figure qui précède, les oxacides sont porteurs de groupes OH. On a pourtant dit en début de ce post que la présence de ce groupe pouvait caractériser une base. Alors qu’est-ce qui différencie la soude NaOH de de l’acide nitrique NO2OH ou de l’acide sulfurique SO2(OH)2 ? A l’état solide la soude est un cristal ionique. Cela signifie que ce cristal est formé de cations Na+ et d’anions OH- maintenus ensemble par des liaisons ioniques. En solution dans l’eau, anions et cations se dissocient. Dans un oxacide la liaison entre l’atome d’oxygène du ou des groupe(s) OH et le minéral est covalente. La dissociation se fait entre l’atome d’oxygène et l’atome d’hydrogène.
Remarque : il existe une troisième catégorie de composés présentant un ou des groupes OH, les alcools. Ils sont généralement amphotères.
En chimie organique, les acides carboxyliques jouent un rôle central (voir le post à ce sujet). Un acide carboxylique est un acide porteur du groupe -COOH. A première vue, la présence du groupe -OH peut paraître surprenante pour un acide. L'acidité des acides carboxyliques s'explique par l’effet inductif de la liaison C=O qui est très polarisée, ce qui fait que le carbone attire les électrons de l'autre atome d’oxygène. Le groupe -OH étant lui aussi polarisé, l'électron de l'atome d’hydrogène est attiré par le carbone « électrophile ». La liaison de l’hydrogène avec le reste de la molécule est donc essentiellement ionique et l’ion H+ peut en être dissocié assez facilement.
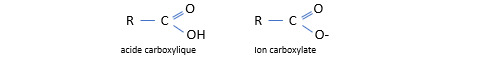
Pour obtenir le nom d’un acide carboxylique, on ajoute le suffixe -oïque au nom de l’hydrocarbure qui constitue l’ossature de sa chaîne carbonée .
HCOOH : acide méthanoïque (aussi appelé acide formique)
CH3-CH2-COOH : acide propanoïque
C6H5-COOH : acide benzoïque
Le nom de l’ion carboxylate correspondant est obtenu en remplaçant le suffixe -oïque par le suffixe -oate. Par exemple, l’ion HCOO- est l’ion méthanoate.
Un acide aminé est acide carboxylique possédant également un groupe amine.
Propriétés des acides
Les acides et les bases sont en général très réactifs. Il convient cependant de faire la distinction entre les réactions acido-basiques et les réactions d’oxydoréduction dans lesquelles des acides (ou des bases) peuvent intervenir. Dans une réaction acido-basique, les réactifs échangent un proton (acide de Brönsted) ou un doublet électronique (acide de Lewis) et le nombre d’oxydoréduction des substances en présence reste inchangé. A contrario, dans une oxydoréduction les réactifs échangent un électron et leur nombre d’oxydation évolue.
Réaction acide/base
La réaction acide/base typique s’écrit comme suit :
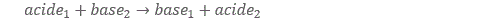
La caractéristique de la réaction acide/base est le transfert de protons. L’acide 1 cède un proton à la base 2 qui le capte et les rôles sont inversés à l’issue de la réaction. La configuration finale est plus stable que la configuration d’origine. On dit qu’il y a eu neutralisation. Le plus souvent, la réaction produit au final un composé peu réactif auquel on a donné le nom de sel, par extension du nom de sel donné au composé chimique NaCl bien connu des cuisiniers/ères.
Reprenons à titre d’exemple l’équation ci-dessus avec une solution de chlorure d’ammonium NH4Cl et de soude NaOH. Il se produit dans cette solution une réaction acide/base dont l’équation est la suivante :

Le transfert de proton apparaît clairement si on décompose cette réaction en deux demi-équations :

La production d’ammoniac se traduit par un dégagement gazeux. L’ion chlorure Cl- et l’ion Na+ quant à eux n’interviennent pas dans ces réactions. Par contre, ils laissent un résidu solide composé de cristaux de chlorure de sodium NaCl après évaporation de l’eau.
Remarque : dans cet exemple, c’est le couple (H2O/OH-) qui intervient dans la deuxième demi-réaction. C’est l’eau en tant que base qui joue le rôle d’accepteur de proton. On dit de l’eau que c’est un solvant protique. Un solvant protique est une solvant capable de céder ou de capter un proton. L’ammoniac, le méthanol ou l’acide sulfurique sont également des solvants protiques.
Comme on l’a vu, une réaction acido-basique conduit souvent à la formation d’un sel mais ce n’est pas systématique. C’est en général le cas lorsqu’on fait réagir un acide et un hydroxyde :

Dans l’exemple cité au début de ce post (réaction de l’ammoniac gazeux avec du chlorure d’hydrogène) on aboutissait également à la production d’un sel, le chlorure d’ammonium bien que l’ammoniac ne soit pas un hydroxyde :

Complexation
Une réaction de complexation est une réaction qui fait intervenir un acide de Lewis avec une base de Lewis. Ce type de réaction se traduit par l’échange d’un doublet d’électron. L’ion Ag+ comme de nombreux cations métalliques, est un acide de Lewis et l’ammoniac est, comme on l’a vu, une base de Lewis. La réaction de l’ion Ag+ avec de l’ammoniac en solution est une réaction de complexation :

Le produit de cette réaction est l’ion diamine argent. Un tel composé est appelé complexe : il associe une base de Lewis et un acide de Lexis.
Oxydoréduction
Les acides peuvent réagir avec les métaux et les oxyder. Ce type de réaction est d’ailleurs une source de corrosion majeure. Dans ce type de réaction, il n’y a pas à proprement parler de transfert de proton. C’est un transfert d’électrons qui se produit, transfert qui est la caractéristique d’une réaction d’oxydoréduction.
Acides non oxydants : l’acide chlorhydrique HCl et l’acide sulfurique H2SO4
Ces deux acides sont des acides forts totalement dissociés mais leur anion (Cl- ou SO42-) n’a pas un potentiel d’oxydation élevé. C’est la raison pour laquelle on les classe dans la catégorie des acides non oxydants. La réaction d’oxydoréduction fait intervenir l'ion hydronium H3O+. C’est cet ion hydronium qui récupère les électrons libérés par les métaux considérés. Ce type de réaction ne se produit qu’avec des métaux très réducteurs comme le fer et le zinc. La réaction se traduit par un dégagement de dihydrogène et la formation d’ions Fe2+ ou Zn2+. Ils n’ont par contre pas d’effet sur des métaux peu réducteurs comme le cuivre ou l’argent. Si on prend l’exemple du fer, l’action du chlorure d’hydrogène se traduit comme suit :

L’ion fer se combine ensuite aux anions chlore pour former du chlorure de fer. On obtient certes l’équation résultante suivante :

mais cette équation est, d’une certaine manière trompeuse. Le dégagement d’hydrogène n’est pas le résultat d’une réaction acide/base mais bien d’une réduction de l’hydrogène libéré par le chlorure d’hydrogène. L’action de l’acide sulfurique sur le magnésium peut s’interprète de la même façon :

Un acide oxydant, l’acide nitrique
L'acide nitrique est un oxydant puissant qui peut oxyder les métaux nobles tels que le cuivre et l’argent. Cette propriété est due cette fois à l'ion nitrate NO3-. L'ion H3O+ n’intervient pas. Lorsqu’on verse de l’acide nitrique sur du zinc par exemple, il y a production d’une abondante vapeur de NO2 à la place du dihydrogène. L'oxydant est donc bien l'anion NO3- dont le nombre d’oxydation est diminué au cours de la réaction. Même chose avec de la poudre d’argent. La présence d’ions argent Ag+ est mise en évidence en diluant dans l’eau le produit obtenu et le faisant précipiter avec du chlorure de sodium (formation de chlorure d'argent).
Cas particulier : l’acide carboxylique
Les acides carboxyliques sont des acides faibles. Ils se dissocient partiellement dans l’eau (pH voisin de 5) :

La chimie des acides carboxylique est riche. Elle est présentée plus en détails dans le post consacré à de type d’acide.
Pour en savoir plus :
post d’introduction à la chimie
post sur les composants élémentaires
pst sur les électrons dans les liaisons covalentes
post sur les liaisons chimiques
post sur les ligands et la complexation
post sur l’oxydoréduction
post sur l’acide carboxylique
post sur l’eau
post sur les sels
post sur les métaux alcalins
post sur la classification périodique des éléments
glossaire de chimie générale
index
#chimie#acide#hydracide#oxacide#hydronium#arrhenius#bronsted#lewis#oxydation#réduction#oxydoréduction#redox#carboxylique#chlorhydrique#sulfurique#nitrique#ph#complexation
0 notes
Text
Tableau périodique des éléments
Dimitri Mendeleïev est un chimiste russe qui vécut et travailla à Saint Pétersbourg au XIXème siècle. Il avait remarqué les similitudes qui existaient entre les propriétés de certains éléments chimiques et il entreprit de les classer en fonction de ces similitudes. Il fallut cependant attendre le début du XXème siècle et l’avènement de la mécanique quantique pour que l’on comprenne la raison profonde de ces similitudes.
La caractéristique principale d’un élément chimique est le nombre de ces protons, que l’on appelle numéro atomique. Les éléments qui ont le même numéro atomique mais dont le noyau comporte un nombre différent de neutrons ont des propriétés chimiques identiques. On les appelle des isotopes. C’est leur degré de stabilité qui les différencie (voir le post sur la structure du noyau atomique). Presque tous les éléments dont le numéro atomique est inférieur ou égal à 80 possède au moins un isotope stable. Les autres isotopes de cet élément sont plus ou moins stables : ils se désintègrent par radioactivité en un élément plus stable au bout d’un certain temps. On appelle demi-vie d’un isotope donné le temps moyen au bout duquel la moitié des noyaux se sont désintégrés.
Nuage électronique
Les propriétés chimiques d’un élément sont déterminées par l’arrangement de son nuage électronique (voir le post sur le nuage électronique). Les électrons d’un atome occupent des états d’énergie quantifiés au sein de ce nuage. L’état quantique d’un électron peut être caractérisé par 3 nombres entiers que l’on appelle par convention n, l et m et un nombre demi-entier, appelé s et qui peut être égal à -1/2 ou +1/2. Ces « nombres quantiques » ne sont pas indépendants les uns des autres. Le premier, n, est le nombre quantique principal :
l est obligatoirement compris entre 0 et n-1,
m est obligatoirement compris entre -l et +l.
On dit des électrons qui ont le même nombre n qu’ils appartiennent à une même couche. Les électrons qui ont le même nombre l appartiennent à une même sous-couche. A ces nombres quantiques correspondent des niveaux d’énergie différents. Les électrons d’un atome ne peuvent pas occuper n’importe quel état. En particulier, deux électrons ne peuvent pas se trouver dans le même état quantique (principe d’exclusion de Pauli). Comme la configuration la plus stable correspond à un état d’énergie minimale à l’échelle de l’atome, les électrons vont donc chercher à remplir les couches successives. Le chimiste Vsevolod Klechkowski a montré que le remplissage se faisait dans l’ordre croissant de la somme n+l puis dans l’ordre croissant de n (règle de Klechkowski).
On repère ces couches et sous-couches par un index composé du nombre n suivi d’une lettre qui caractérise la sous-couche l (s, p, d, f par ordre croissant). Par exemple, l’index 3p caractérise la sous-couche telle que n = 3 et l = 1. Cette sous-couche peut comporter 6 électrons (voir la figure ci-dessous).

Valence et règle de l’octet
Seuls les électrons appartenant à la couche de nombre quantique n le plus élevé participent aux propriétés physico-chimiques de l’élément considéré. Les couches inférieures correspondent à des orbitales plus proches du noyau et les électrons qui les occupent sont piégés dans un puits de potentiel plus profond. De manière imagée, on peut dire que les électrons dont le nombre n est le plus élevé sont « à la périphérie » du nuage électronique. Leur énergie de liaison est plus faible que celle des autres atomes. Ils sont, d’une certaine manière, disponibles pour établir des liaisons chimiques ou pour intervenir dans des réactions chimiques. On appelle bande de valence la couche de nombre n le plus élevé. On appelle valence le nombre maximal de liaisons chimiques qu’un éléments peut former.
Les éléments dont la bande de valence présente une organisation similaire ont des propriétés physico-chimiques voisines. Prenons deux exemples : celui du carbone et du silicium. Le carbone a le numéro atomique 6, il a donc 6 électrons. Sa couche n = 1 est remplie ainsi que la sous-couche 2s. Par contre la sous-couche 2p ne comporte que 2 électrons alors qu’elle pourrait en comporter 6. Le silicium a quant à lui le numéro atomique 14. Ses couches n = 1 et n = 2 sont remplies, sa sous-couche 3s est remplie mais sa couche 3p ne l’est que partiellement. Elle aussi ne comporte que 2 électrons alors qu’elle pourrait en comporter quatre de plus. C’est cette similitude qui explique que ces deux éléments présentent des propriétés voisines.
Tous deux sont tétravalents. ils peuvent mobiliser quatre électrons de valence, deux sur la sous-couche -s et deux sur la sous-couche -p. Ils ont par ailleurs quatre orbitales inoccupées sur ladite sous-couche -p. Ils peuvent donc former simultanément quatre liaisons chimiques. C’est le cas avec l’oxygène dont le numéro atomique est 8. La couche n = 1 de l’oxygène est remplie, sa sous-couche 2s également mais sa sous-couche 2p ne comporte que 4 électrons alors qu’elle pourrait en comporter 6. L’oxygène est donc divalent. Récapitulons : le carbone a 4 électrons sur sa bande de valence, l’oxygène en a deux. Le carbone a 4 places « disponibles » sur sa sous-couche 2p et l’oxygène en a deux. Cela permet au carbone et à l’oxygène de former la molécule de dioxyde de carbone CO2 au sein de laquelle le carbone entretient une double liaison avec chacun des atomes d’oxygène (O=C=O). Il en va de même pour le silicium qui est oxydé par l’oxygène pour former la molécule SiO2, la silice, qui a la même structure (O=S=O) que le CO2. On pourrait citer également l’exemple du méthane (CH4) et du silane (SiH4). Cette fois on a affaire avec l’hydrogène à un atome univalent : un électron sur la couche 1s et une orbitale libre. On pourra remarquer d’ailleurs que le carbone et le silicium cristallisent tous deux sous la forme d’un cube à face centrée.
Remarque : la sous-couche 3d du silicium est entièrement vide. Mais dans ce cas, si l’on se réfère à la règle de Klechkowski (voir plus haut) on saute un niveau d’énergie puisque n + l = 5 > n + 1.
Prenons un autre exemple, celui de l’oxygène (divalent) avec l’hydrogène (univalent). L’oxygène et l’hydrogène se combinent pour former la molécule d’eau H2O. L’atome d’azote est quant à lui trivalent : numéro atomique 7, couche n = 1 et 2s remplies, sous-couche 2p comportant 3 électrons et présentant 3 orbitales libres. L’azote peut se combiner avec trois atomes d’hydrogène pour former de l’ammoniac NH3. On ne sera pas étonné de constater que le phosphore (numéro atomique 15, sous-couches 1s, 2p, 3s remplies, sous-couche 3p ne comportant que 3 électrons) peut lui aussi se combiner avec l’hydrogène pour former de la phosphine PH3 présentant une structure tétraédrique similaire à celle de l’ammoniac.
Ces exemples illustrent une règle très générale : la règle de l’octet. Tous les éléments chimiques dont la bande de valence est de type -s ou -p (c’est-à-dire telle que l = 0 ou 1) à l’exception de l’hydrogène, de l’hélium, du lithium et du béryllium) ont tendance à se combiner de façon à remplir ces deux sous-couches. C’est ce qui lui vaut ce nom de règle de l’octet, les états disponibles étant au nombre de 8 (2+6).
Tableau de classification périodique
Venons-en au tableau proprement dit. Il comporte 7 lignes et 18 colonnes auxquelles s’ajoutent deux lignes supplémentaires.

Le remplissage des lignes suit la règle de Klechkowski.
1s
2s – 2p
3s – 3p
4s – 3d – 4p
5s – 4d – 5p
6s – 4f – 5d – 6p
7s – 5f – 6d – 7p
Les lignes supplémentaires (8 et 9) correspondent aux sous-couches 4f et 5f qui s’insèrent dans les lignes 6 et 7. A une colonne correspond un nombre d’électrons donné dans une sous-couche de même nombre quantique l. Ce tableau met bien en évidence les grandes classes d’éléments chimiques :
La colonne repérée en rouge (un électron dans la bande de valence) est celle des éléments alcalins. Ils réagissent avec les halogènes (voir plus loin) ainsi qu’avec l’eau pour donner des hydroxydes (bases fortes).
La colonne brun-clair est celle des alcalino-terreux. Ils réagissent aussi avec les halogènes mais pas avec l’eau.
De l’autre côté du tableau (colonne bleu-clair) on trouve les gaz nobles. Leur bande de valence est complètement remplie et ils ne réagissent avec aucun autre élément chimique. Ils n’existent que sous la forme de gaz monoatomique.
A gauche de cette colonne, la colonne jaune est celle des halogènes. Ils forment des acides forts avec l’hydrogène (comme l’acide chlorhydrique HCl) et ils forment des composés ioniques avec les alcalins (comme le chlorure de sodium NaCl) et les alcalino-terreux (comme le chlorure de calcium CaCl2).
Les éléments de la colonne 15 sont appelés pnictogènes et ceux de la colonne 16 chalcogènes. Ces dénominations sont peu utilisées mais elles peuvent toujours servir au scrabble ou pour les mots croisés. Ils présentent néanmoins une réelle parenté chimique :
Colonne 16 : oxygène, soufre, sélénium, tellure, polonium
Colonne 15 : azote, phosphore, arsenic, antimoine, bismuth
Pour la colonne 14 (carbone, silicium, germanium, étain, plomb), la situation est plus contrastée. Le germanium cristallise comme le carbone et le silicium. Silicium et germanium sont tous deux utilisés pour réaliser des semi-conducteurs. Par contre, en ce qui concerne l’étain et le plomb, c’est leur caractère métallique qui domine.
Les éléments de la ligne 8 sont appelés lanthanides (sous-couche 4f). On leur donne également le nom de terres rares. Ceux de la ligne 9 sont des actinides. Ils présentent des propriétés particulières.
Les couleurs utilisées dans le milieu du tableau correspondent à un autre type de classification. La couleur chair correspond aux éléments dits métalliques (plus exactement métaux de transition). Les éléments en gris foncés sont des métaux pauvres. Ceux en kaki sont des métalloïdes. La répartition entre ces catégories varie selon les auteurs, leur caractère métallique étant plus ou moins prononcé.
Pour en savoir plus :
post sur la structure du noyau atomique
post sur le nuage électronique
post sur les liaisons covalentes
post sur les liaisons chimiques
post sur la valence
post sur la cohésion de la matière
post sur les éléments chimiques
post sur les halogènes
post sur les métaux alcalins et alcalino-terreux
post sur les métaux de transition
post sur la radioactivité
glossaire de chimie générale
index
#chimie#tableau périodique#Mendeleiev#alcalin#halogène#métal#électron#proton#radioactivité#isotope#liaison chimique#matière#élément#nuage électronique#actinide#lanthanide#pnictogène#chalcogène#carbone#silicium#oxygène#hydrogène
0 notes