#liaison covalente
Explore tagged Tumblr posts
Visit Tumblr Blog
Explore Tumblr blogs with no restrictions, modern design and the best experience.
Last Seen Tumblr Blogs





Fun Fact
Tumblr has 411 employees.
Text
H2O, une molécule si particulière
La molécule d’eau a des propriétés très particulières sans lesquelles la vie n’existerait sans doute pas, du moins sous la forme qu’on lui connaît. Quelles sont ces propriétés « miracle » ?
Une température de fusion et une température d’ébullition raisonnables
L’eau reste à l’état liquide dans une plage de température relativement étendue : de 0°C à 100°C. Ce n’est pas le cas des autres composés les plus abondants dans l’Univers. La température de fusion de l’ammoniac (NH3) est de -77,7° C, sa température d’ébullition de -33,3° C. Pour le méthane (CH4) ces températures sont respectivement de -182,5° C et -161,5° C. Les plages correspondantes sont nettement plus étroites et les températures plus faibles. Or, une plage étendue garantit le maintien de l’eau à l’état liquide malgré de fortes amplitudes thermiques. Les océans jouant le rôle de régulateur thermique, c’est un facteur clef pour le développement de la vie. Une température d’ébullition trop basse pourrait d’ailleurs s’avérer rédhibitoire : cela signifierait que l’eau ne pourrait se maintenir à l’état liquide qu’à une distance plus grande du Soleil… D’où un ensoleillement moindre (et on sait qu’il joue un rôle déterminant dans la photosynthèse) et des réactions chimiques bien plus lente (un handicap pour le métabolisme).
La masse volumique de la glace
La masse volumique de la glace est inférieure à celle de l’eau à l’état liquide. La glace flotte ! Lorsque la température de l’air descend en dessous de zéro, l’eau gèle en surface et la glace ne coule pas. Il se forme au contraire une couche de glace qui isole l’eau qui se trouve en dessous du froid ambiant. Celle-ci reste à une température voisine de zéro. Si la glace était plus dense que l’eau liquide, les rivières et les lacs gèleraient en profondeur. Or, on suppose que la vie à commencé à se développer dans l’eau. Si toutes les étendues d’eau étaient prises dans les glaces (lors des périodes glaciaires par exemple), elle n’aurait sans doute pas pu dépasser le stade de microorganismes peu différenciés.
Le cycle de l’eau
La vapeur d’eau est plus légère que l’air. L’air humide s’élève et forme des nuages en se refroidissant. Le vent les emporte et les précipitations arrosent la terre ferme et les reliefs.
Les propriétés de solvant de l’eau
L’eau est un solvant pour certains composés organiques et pas pour d’autres. Cette propriété joue un rôle déterminant dans la formation des cellules et dans leur métabolisme. La dissolution du dioxyde de carbone par l’eau a également joué un rôle important pour séquestrer le CO2 abondant dans l’atmosphère pendant les premières centaines de millions d’années.
Je vous propose un voyage dans un verre d’eau pour essayer de comprendre l’origine de ces propriétés tout à fait exceptionnelles.
La molécule d’eau
La molécule d’eau est constituée d’un atome d’oxygène, de masse molaire égale à 16 g, et de deux atomes d’hydrogène, de masse molaire égale à 1 g. La masse molaire de la molécule d’eau vaut donc 18 g. Elle est notablement inférieure à la masse molaire moyenne de l’air (29 g). L’air est en effet composé principalement de diazote (N2, 28 g) et de dioxygène (O2, 32 g). Cette propriété de l’eau joue un rôle déterminant dans le régime hydrographique et celui des précipitations. L’eau qui s’évapore à la surface des océans humidifie l’air. L’air humide est plus léger que l’air sec du fait de sa teneur en vapeur d’eau. Il s’élève et se refroidit : la température diminue d’un degré tous les 150 m. La condensation partielle de l’humidité forme des nuages qui sont transportés au loin par les vents dominants. Ces nuages donnent des précipitations qui arrosent les reliefs. L’eau de pluie (ou la neige fondue) s’infiltre dans le sol et alimente les sources. L’eau des rivières draine alluvions et sédiments, participant ainsi à la fertilité des sols, avant de rejoindre l’océan.
La boucle est bouclée… tout cela grâce à la faible masse molaire de la molécule d’eau��!
Oxygène et hydrogène…
Revenons à nos moutons. Un atome d’oxygène pour deux atomes d’hydrogène : H2O. L’oxygène est le troisième élément le plus abondant dans l’Univers après l’hydrogène et l’hélium. Son numéro atomique est 8. Il comporte donc 8 électrons. Ceux-ci sont répartis de la manière suivante : deux électrons occupent la couche 1s (voir le post sur les électrons), deux la sous-couche 2s et quatre la sous-couche 2p. Parmi ces derniers, deux sont non appariés (ils sont seuls sur leur orbitale). Ceci permet à l’oxygène de nouer deux liaisons covalentes (voir le post sur les bases de la chimie).
L’hydrogène est l’élément le plus abondant dans l’Univers. Il n’a qu’un seul électron sur sa couche 1s. Dans la molécule H2O, les deux atomes d’hydrogène mettent en commun leur électron avec les deux électrons non appariés de l’oxygène pour former deux liaisons covalentes simples. Les orbitales 2p sont orthogonales entre elles, on pourrait s’attendre à ce que la chaîne H-O-H soit à angle droit mais la répulsion coulombienne écarte les deux branches. L’angle résultant vaut 104,5 degrés.
Répulsion coulombienne ? N’y a-t-il pas neutralité électrique des atomes au sein de la molécule ? Pas tout à fait. L’oxygène a un potentiel électronégatif très fort et il « tire la couverture électronique » vers lui. De ce fait, la molécule d’eau présente un moment dipolaire. L’atome d’oxygène porte une charge négative et les atomes d’hydrogène une demi-charge positive.
Signalons enfin que l’atome d’oxygène comporte deux doublets électroniques non liants (les électrons appariés de la sous-couche 2s et ceux de l’orbitale de la sous-couche 2p non impliquée dans les liaisons covalentes). Ces doublets participent au caractère électronégatif de l’atome d’oxygène.
Liaison hydrogène
Présence d’atomes d’hydrogène porteurs d’une charge positive, existence de doublets non liants : la molécule d’eau peut nouer des liaisons hydrogène avec d’autres molécules d’eau. Et elle ne s’en prive pas !
La liaison hydrogène est une liaison directionnelle de nature principalement électrostatique. Elle s’établit entre un atome d’hydrogène et un atome d’oxygène, d’azote ou de fluor, ces trois éléments étant connus pour avoir un fort potentiel électronégatif. L’énergie de liaison d’un pont hydrogène est moins élevée que dans le cas d’une liaison covalente mais plus fort que celle des liaisons électrostatiques classiques. En termes savants, on caractérise cette liaison en disant qu’elle relie une molécule « donneuse » et une molécules « accepteuse » :
la molécule donneuse est un composé possédant un atome d’hydrogène porteur d’une charge positive (qui peut être partielle),
la molécule accepteuse comporte un atome porteur d’un doublet non liant.
La molécule d’eau répond à des deux critères.

Cette liaison est directionnelle : la direction est imposée par la forme de la molécule. Elle s’établit dans l’eau à l’état liquide et à l’état solide. C’est la liaison hydrogène qui structure les cristaux de glace et cette structure, relativement aérée, qui fait que la densité de la glace est plus faible que celle de l’eau liquide. Le caractère directionnel de cette liaison impose aux cristaux de glace une forme particulière qui va à l’encontre de leur compacité. En contrepartie, l’énergie de cette liaison permet à la glace de rester à l’état solide à une température plus élevée que le méthane et l’ammoniac qui ont pourtant des masses molaires voisines (18 g pour l’eau, 17 pour l’ammoniac et 16 pour le méthane). Pour le méthane, la raison en est évidente. Du fait de se nature tétraédrique (avec le carbone au centre), la molécule est strictement apolaire. Le méthane à l’état solide forme des cristaux moléculaires dont les éléments sont faiblement liés entre eux. Pour l’ammoniac c’est plus subtil. L’ammoniac et une molécule polaire mais son moment dipolaire est plus faible que celui de l’eau. La position de l’atome d’azote oscille entre « les deux sommets » de la pyramide formée avec les trois atomes d’hydrogène : celui du dessus et celui du dessous. Ce moment plus faible rend moins solide les liaisons au sein du cristal d’ammoniac à l’état solide.
Les ponts hydrogène jouent également un rôle dans l’eau liquide. Ils créent une certaine forme « d’adhérence » entre les molécules. Une molécule d’eau ne se déplace jamais seule... Ces liaisons hydrogène « alourdissant » les molécules d’eau et entravent le passage à l’état gazeux. La liaison hydrogène nous permet donc de faire d’une pierre deux coups et d’expliquer la plage de température dans laquelle l’eau reste à l’état liquide et la légèreté de la masse volumique de la glace.
Les propriétés de solvant de l’eau
Le caractère dipolaire de la molécule d’eau lui confère d’excellentes propriétés de solvant. L’eau dissocie facilement les cristaux ioniques comme le chlorure de sodium (NaCl). Les cristaux ioniques sont des cristaux formés de molécules composées d’un cation (un ion positif) et d’un anion (un ion positif) de charge opposée. (Dans la vie courante on les appelle tout simplement des sels.)
Mais plus généralement l’eau dissout les composés chimiques polaires, c’est-à-dire présentant eux aussi un moment dipolaire. Ce moment leur permet de créer des liaisons hydrogène avec l’eau. On dit de ces composés qu’ils sont hydrophiles. Des molécules organiques comme les amines et les alcools sont hydrophiles. Les cétones (un groupe carbonyle -C=O) le sont également.

Les composés apolaires (dépourvu de moment polaire) sont hydrophobes. Ils repoussent l’eau, n’ayant pas la possibilité de créer une liaison hydrogène ni même une liaison électrostatique simple avec elle. Les alcanes (hydrocarbures saturés : méthane, éthane, propane…) sont hydrophobes.
Certains composés organiques complexes sont amphiphiles : ils comportent un groupe fonctionnel hydrophile et un groupe hydrophobe (généralement une longue chaîne carbonée). En présence d’eau, ces composés ont des propriétés tensioactives : ils créent une interface séparant le milieu aqueux (dans lequel peuvent être dissous des composés hydrophiles) du milieu non aqueux. La partie hydrophile de ces composés est tournée vers le milieu aqueux, la partie hydrophobe formant quant à elle une sorte de membrane. Cette membrane peut se refermer sur elle-même, formant ainsi une petite enveloppe plus ou moins perméable. Les composés amphiphiles jouent donc un rôle essentiel dans la chimie du vivant ! La paroi des cellules vivantes est composée de composés amphiphiles.
Les composés amphiphiles permettent par ailleurs de mélanger dans la même solution aqueuse des molécules hydrophiles (des ions par exemple) et des groupes fonctionnels hydrophobes, ce qui rend possible des réactions autrement difficilement concevables. Encore un atout pour la chimie du vivant.
Remarque : le savon est le plus connu des composés amphiphiles. Ses propriétés tensioactives sont à la base des bulles de savon, de très fines membranes emprisonnant une mince couche d’eau.
Eau, acides et bases
Un acide dissous dans l’eau libère des ions hydronium H3O+ :

Une base dissoute dans l’eau libère des ions hydroxyde OH- :

L’ion hydronium et l’ion hydroxyde sont à la base de la plupart des propriétés des acides et des bases. A l’état naturel, l’eau comporte une petite dose de ces ions. Elle est produite par autoprotolyse de l’eau :

Dans l’eau « pure », il y a équilibre entre les concentrations de ces deux ions.

L’inverse du logarithme en base 10 de la concentration en ions hydronium caractérise l’acidité d’une solution (pH de cette solution). Le pH de l’eau pure est donc égal à 7. Un pH inférieur à 7 indique qu’il y a plus d’ions hydronium que d’ions hydroxyde. La solution est acide. A l’opposé, un pH supérieur à 7 indique que la solution est basique.
Les réactions acido-basiques sont à la base d’une multitude de réactions dans notre environnement.
H2O et CO2
La molécule de dioxyde de carbone est constituée d’un atome de carbone qui entretient deux doubles liaisons covalentes avec des atomes d’oxygène (O=C=O). C’est une molécule très stable. Fort heureusement pour nous, le CO2 est soluble dans l’eau :

La molécule H2CO3 est un acide, l’acide carbonique. C’est l’acide carbonique qui est à l’origine des pluies acides. La présence d’acide carbonique dans l’eau se traduit par un équilibre entre acide, ion hydrogénocarbonate et ion carbonate :

La présence de silicates en suspension dans l’eau favorise elle aussi la production d’ions hydrogénocarbonates :

(Cette réaction est valable pour tout type de silicates comportant des atomes de calcium.)
La dissolution du dioxyde de carbone dans l’eau joue un rôle important dans le cycle du carbone. L’eau des océans s’évapore, augmentant le taux d’humidité dans l’air. Le CO2 présent dans l’atmosphère se dissout dans les gouttes d’eau en suspension. (Du CO2 est également dissous directement dans l’eau à la surface des océans.) Les pluies acides créent un ruissellement qui favorise la formation d’ions hydrogénocarbonates et de silice. L’eau chargée d’ions hydrogénocarbonates (et dans une moindre mesure d’ions carbonates) ainsi que d’ions calcium rejoint les océans.
Que deviennent ces ions ? Il y a trois possibilités de recyclage de ces ions.
La première : les ions calcium Ca++ précipitent avec des ions hydrogénocarbonates et carbonates :

Le carbonate de calcium CaCO3 sédimente au fond de l’océan. Au cours des premières dizaines (voire centaines) de millions d’années, cette sédimentation a conduit à une séquestration massive de CO2 dont la teneur a chuté de manière drastique dans l’atmosphère.
La seconde : les ions Ca++ et HCO3- sont absorbés par les organismes marins pour secréter leur squelette et leur coquille. Lorsqu’ils meurent, squelettes et coquilles rejoignent le fond de l’océan et sédimentent à leur tour. Ce mode de séquestration du CO2 est, de nos jours, largement prépondérant par rapport au mode abiotique décrit ci-dessus.
La troisième, l’augmentation de la concentration en acide carbonique dans l’eau entraîne la libération de CO2 qui s’évapore à la surface des océans :

C’est la réaction inverse de celle qui conduit à la dissolution du CO2 dans l’eau.
Terminus, tout le monde descend…
C’est la fin de notre voyage dans un verre d’eau. Comme on le voit, les propriétés de l’eau ont joué et continuent de jouer un rôle déterminant dans la chimie du vivant. Aucun autre composé chimique connu ne rassemble autant de propriétés favorisant le développement de la vie. Mais d’où vient l’eau ? Etait-elle contenue dans les astéroïdes qui ont formé la proto-terre ? A-t-elle été apportée ultérieurement par des comètes lors d’un épisode de bombardement tardif ? A-t-elle une origine double ? La question n’est pas tranchée. Une meilleure connaissance de la composition isotopique de l’eau contenue dans les comètes permettra de valider ou d’invalider la deuxième hypothèse. Une chose est sûre, la Terre s’est formée en deçà de la ligne des glaces. Contrairement à ce qui s’est passé pour les planètes gazeuses (et leurs satellites) elle n’a pas pu accréter de grandes quantités d’eau sous forme de glace.
Une autre conclusion s’impose : la Terre a eu beaucoup de chance de conserver son eau. Au cours des premières centaines de millions d’années, la planète Mars possédait beaucoup d’eau à l’état liquide. Mais Mars n’a pas pu garder son atmosphère. En l’absence de champ magnétique, les vents solaires l’en ont progressivement dépouillé. La pression a alors chuté sous un seuil qui ne permettait plus à l’eau de rester à l’état liquide. Elle s’est évaporée. La photodissociation des molécules d’eau en altitude a fait le reste. Les atomes d’hydrogène se sont échappés dans l’espace. Leur faible masse leur ont permis d’acquérir rapidement une vitesse supérieure à la vitesse de libération du fait des UV et des collisions avec les particules du vent solaire. L’eau liquide a disparu de la surface de Mars.
Vénus a connu un sort différent mais qui a abouti au même résultat. Vénus est la sœur jumelle de la Terre mais elle n’est qu’à 0,7 UA du Soleil. L’effet du rayonnement y est donc beaucoup plus important. L’atmosphère de Vénus était sans doute composée des mêmes ingrédients que l’atmosphère terrestre. Il y régnait donc un puissant effet de serre et la température ambiante était beaucoup plus élevée que sur Terre. Idem pour le taux d’humidité. On suppose que ces différences se traduisaient par des mouvements de convexion de grande ampleur qui faisaient monter la vapeur d’eau jusqu’à la limite de la troposphère. Là, les molécules d’eau ont été photodissociées par le rayonnement solaire et l’hydrogène a échappé à l’attraction vénusienne comme cela s’est passé sur Mars. Il s’en est suivi une perte continue en eau. L’atmosphère s’est asséchée. Le mécanisme de séquestration du CO2 qui a bien fonctionné sur Terre a échoué sur Vénus. L’effet de serre s’est emballé et la température au sol a dépassé le point critique de l’eau (374° C). Les océans se sont évaporés complètement...
L’eau est bien un composé miracle, dans tous les sens du terme.
Pour en savoir plus :
post sur la formation de la Terre
post sur les bases de la chimie
post sur les composants principaux de la vie sur Terre
post sur l’abondance des éléments dans le corps humain
post sur le dioxyde de carbone
post sur les électrons et la liaison covalente
post sur les liaisons chimiques
post sur la cohésion de la matière
post sur les acides et les bases
post sur le point triple de l’eau
post sur le point de rosée
glossaire de chimie générale
index
#eau#h2o#co2#physique#terre#oxygène#hydrogène#chimie#liaison covalente#liaison hydrogène#azote#mars#vénus#hydronium#hydroxyde#carbonate#hydrogénocarbonate#ammoniac#méthane#hydrophile#hydrophobe#amphiphile#chimie organique#glace#solvant
1 note
·
View note
Text
biochimie, inhibiteurs
L'activité d'une enzyme peut être modulée par d’autres molécules :
un inhibiteur est une molécule qui diminue l'activité d'une enzyme,tandis qu'un
activateur l'accélère ; de nombreux médicaments mais aussi des poisons sont des inhibiteurs enzymatiques.
Un inhibiteur est une molécule, généralement de petite taille, qui lorsqu’elle se lie à une enzyme, en diminue l’activité catalytique.
Elle peut
⚙️empêcher la fixation du substrat en se liant à sa place dans le site actif, ou bien encore
⚙️provoquer une déformation plus ou moins étendue de la structure tridimensionnelle de l'enzyme ne permettant plus d’assurer la catalyse de la réaction.
L’inhibition peut être réversible ou bien irréversible.
Les inhibiteurs dits réversibles se lient aux enzymes par des liaisons non covalentes, de faible énergie, comme des liaisons hydrogène, des liaisons ioniques ou encore des interactions hydrophobes.
L’ensemble de ces liaisons permet d’établir une association plus ou moins forte et plus ou moins spécifique entre l’enzyme et l’inhibiteur.
(tips pour exam) :
Déformation structure 3D enzyme
Réversible liaison h ionique
Permettent liaison enzyme donc modif 3D
Compétitif - ressemblance structurale
Affinité apparente ⬇️vitesse Ma⬆️
Incompéte
Affinité apparente ⬆️vitesse Max⬇️
0 notes
Text
Disneyland star wars sabre de lumière
CDE Fashion une vaste gamme de bijoux pour les détaillants dans le courant Bijoux place.Beach des éléments tels que des étoiles de mer, coquillages et rocks. Il peut être très stressant préparent une proposition parce que vous voulez Statue du panther noir de disney seulement le faire une fois. Les hommes qui portent la barbe à travailler lundi sera renvoyé chez lui sans paie et peuvent être congédiés s'ils ne retournent pas rasé de près par jeudi.agent trouve le coffre Passman dans la cour arrière de la chambre sur cour Buckspurla police.Nous employons environ 12 personnes, principalement des femmes,. Ce bijoux apparaît rarement dans les magasins, implique rarement de joyaux et est rarement, voire jamais, pris par l'chatterers de style commercial. 'La pierre que nous appelonsdisney est un matériau célèbre avec superlatifs qualités physiques, dont la plupart proviennent de la liaison covalente forte entre ses atomes. Ses amis sont sûrs de se pâmer quand ils entendent l'histoire. La monnaie des États-Unis peuvent manquer de bijoux boutique commodités comme miroirs fastueuse et parfumé air, Disney Déguisement Death Trooper Pour Enfants, Rogue One : a Star Wars Story à la mode récent mais les bijoux vendu il y est de qualité supérieure. Effrayant, passionnant et de la beauté qui vient avec la tension entre ces feelings. Depuis de nombreuses années auparavant, il avait travaillé pour housse maker confortables Manufactu site officiel chanel d'abord à Baltimore et plus tard dans le Carolinas. également envisager un brassard osseuse et une série de Disney Poupée Classique Prince Philippe, La Belle Au Bois Dormant dernière mode du lobe d'argent. Chaque direction du service préféré son propre gigoteuse disneyemblème, de sorte que les disney la chaine ca avait des ailes pour la Force aérienne, un aigle sur un globe et point d'ancrage pour le Corps des Marines, une ancre et les initiales USN pour l marine, ou un aigle pour l'armée. Marshall est la ligne du premier compte.Le vendredi, QVC était déjà sur l'air avec une réplique d'une Disney Puzzle 64 Pièces La Reine Des Neiges : Une Fête Givrée Vente Chaleur par Lane que vendu aux enchères le jeudi.Natural sont formés avec des températures élevées etune profondeur d'environ 140190 kilomètres (87118 mi) dans le manteau de la Terre de pression. Vashi handpicks world disney france pour leur feu, la brillance et de l'éclat (ainsi que les 4C - coupe, couleur, clarté et carat) «en fait, il est parce que nous disons non, disent-ils yes'After cette première sélection, un orfèvre (joué par Alex. www.disneystuffs.com
0 notes
Text
10 Bienfaits de l’Eau – qu’est Ce que c’est, et les Propriétés
Ce qui est dans l’eau que nous prenons sur une base quotidienne? Quelles sont les propriétés pour en faire de l’eau-elle si importante? Et quels sont les avantages de l’eau, vous pouvez vous attendre à votre santé et votre forme? Vous verrez que tout ce qui suit, combien en prendre, et beaucoup de conseils.
L’eau est un composé inorganique) se compose de deux atomes d’hydrogène et un d’oxygène liés par des liaisons du type des liaisons covalentes. À température ambiante, environ 25 degrés C, et il est dans une forme liquide. Et ce n’est pas une coïncidence que la Terre qui est également désigné comme une “Planète Bleue” – environ 70% de la surface terrestre est recouverte par l’eau. La quantité totale d’eau sur terre, près de 97% est dans les océans et les mers, environ 2% de la elle est solidifiée pour former les glaciers, et le 1% et le reste se trouve dans les rivières, les lacs, et le métro.
La quantité d’eau douce disponible est très limité, donc si vous prenez en compte le nombre total de personnes dans le monde, et est aussi inégalement réparties entre les continents. Dans une étude menée par l’Université de Columbia (etats-unis)), a montré une corrélation entre la rareté de l’eau et de l’apparition de guerres. Dans la période de 1950 à 2004, les chances de l’éventualité d’une guerre civile, a pris son essor dans 90 pays tropicaux qui sont touchés par le phénomène climatique connu comme El Niño, qui, de temps à autre pour promouvoir l’augmentation de la température et une diminution de la quantité de précipitations dans le monde.
Maintenant, nous allons apprendre à connaître les bienfaits de l’eau, de son importance et de certaines de ses propriétés.
Ce qui est dans l’eau?
Où est l’eau c’est la vie. Il y a une hypothèse sur l’origine de la vie, la plus fondamentale et la plus primitive, “l’organisation moléculaire semblable à une cellule dans la coacervados, il a surgi dans l’environnement aquatique. C’est comme de l’eau, de solvant ou d’un métabolite, dans l’eau, il est essentiel pour n’importe quel organisme vivant.
Environ 60% à 70% du corps humain est composé d’eau, et 73% du cerveau et du cœur, 83% de cancer du poumon et de 64% de la peau, 79% de la les reins et les muscles, 90% du sang, et 31% des os de l’eau. Toutes les réactions chimiques du métabolisme se produire dans le milieu de l’eau. Bien qu’il ne se passe sur l’eau à la fin du processus pour la synthèse de l’énergie (la respiration aérobie), il n’est pas nécessairement par la prise d’eau que nous avons mis de nouveau ce que nous avons perdu par les fluides corporels tels que la sueur et l’urine.
Et en parlant de la sueur et l’urine, nous allons mettre l’accent sur deux autres aspects essentiels de l’eau-qui est, par le biais de l’élimination de la sueur, ou de la transpiration, l’eau aide à maintenir et à contrôler la température du corps, grâce à la sécrétion de l’urine, nous avons été en mesure de se débarrasser des déchets et des toxines résultant du métabolisme de la nourriture, des médicaments, et une foule d’autres composés xénobiotiques.
En présence de l’eau dans les articulations, et il est vital pour la lubrification de la même chose. L’eau sera également aider à donner une protection pour les organes, les personnes de santé fragile et de la vie, tels que le cerveau et la moelle épinière. L’eau du placenta (placenta) sera également amortir le possible choc et le traumatisme pour le fœtus.
Voir aussi:
Les propriétés de l’Eau
L’eau pure ne présente pas de calories, ne contiennent pas de glucides, de protéines ou de la graisse.
L’eau que nous buvons peut avoir absorbé certains des minéraux et des avantages pour la santé humaine. Le calcium et le magnésium, par exemple, peut être utilisé pour les os et le système cardiovasculaire. Le Sodium est un électrolyte essentiel pour le milieu extracellulaire, ce qui est fondamental pour le cerveau et les cellules nerveuses, et est impliqué dans la régulation de la pression artérielle. Et oui, il est facilement submergé par une production excessive de sueur. Le couvercle est en place pour un agent, un antioxydant, ce qui est important pour l’utilisation du fer, et le système cardio-vasculaire. Le sélénium a également des propriétés anti-oxydantes et aide le système immunitaire. Déjà, le potassium est très demandé dans les différents processus métaboliques, bien qu’il ne se trouve pas en grande quantité dans l’eau potable.
Il est important de frisarmos que de la quantité et de la qualité de ces spécimens dépendra du type de l’eau dans la zone où elle a été prise. La commune de l’eau que nous buvons de l’eau du robinet, en particulier la soi-disant “difficile » de l’eau, par exemple, le calcium et le magnésium. Le type de minéral, en plus de ces éléments, vous pouvez prendre le sodium.
Le fluorure est ajouté à l’approvisionnement en eau de nombreuses villes, comme une mesure préventive pour des caries dentaires.
Les bienfaits de l’Eau
Les bienfaits de l’eau sont nombreuses et essentielles à la vie, à la santé et au bon ordre. Nous allons voir quelques-uns des suivants.
1) Boire de l’eau aide à mincir
Dans une étude menée par l’Université de Birmingham, au royaume-Uni, a montré que boire de l’eau avant les repas vous aider à perdre du poids. Des 84 les adultes qui sont obèses, les participants à l’étude ont reçu des directives générales sur la façon de mincir, et ils ont été divisés en deux groupes. Dans l’un d’eux, et qu’ils auraient à utiliser votre imagination, et d’imaginer que leurs estomacs sont pleins avant le repas; dans le groupe de contrôle, les volontaires de boire environ 500 mL d’eau, 30 minutes avant un repas. Les chercheurs ont suivi tous les participants dans le cadre de l’étude, qui a duré 12 semaines, et de surveiller votre poids, l’activité physique, et de l’urine d’entre eux (dans ce cas, afin d’avoir un contrôle sur ceux qui étaient en train de boire plus d’eau. Les résultats ont été très satisfaisants pour ceux qui ont pris l’eau avant les repas emagreceram la plupart des gens de l’autre groupe (environ 1,4 Kg) et plus); et ceux qui ont été effectivement prises dans le (500 ml) ont eu une perte de poids plus prononcé (plus ou moins, à 4, à 3 Lbs.)
Boire de l’eau non seulement avant, mais aussi dans l’intervalle des repas, augmente le sentiment de satiété, ainsi que d’améliorer votre digestion et prévenir la constipation. Les avantages de l’eau par rapport à la perte de poids est aussi important, et il serait un grand encouragement pour les gens à boire plus d’eau chaque jour.
(2) L’eau est essentielle à la croissance musculaire.
L’eau n’est plus nécessaire, ou plus de protéines, de graisses et de glucides dans le bâtiment, et le développement des muscles. La déshydratation altère le processus digestif et, par conséquent, l’absorption de tous les autres nutriments dans le corps.
Dans un article publié dans le » Journal of Force et Conditionnement de la Recherche a démontré qu’une perte de seulement environ 1,5% de l’eau, il favorise une réduction de la force musculaire. La force musculaire est essentiel pour la croissance musculaire, comme c’est le facteur qui vous permet d’optimiser vos séances d’entraînement, augmenter le poids.
Pour la prévention des blessures du muscle est encore un autre avantage offert par l’eau. En fonction de la quantité d’eau présente à l’intérieur de la fibre, car il peut être, ou pas à la dégradation du tissu musculaire. Des études ont montré qu’une diminution de la quantité d’eau dans notre corps fournit pour le rétrécissement des cellules, et la dégradation de la protéine.
N’oubliez pas de boire de l’eau avant et pendant votre séance d’entraînement. Vous garder hydraté est essentiel pour la récupération et la croissance musculaire.
3) l’eau contribue au bon fonctionnement des reins
Tout cela est qu’il est métabolisé dans le corps, il est pris dans le flux sanguin. Dans les reins, plus précisément, les néphrons, sont responsables pour le filtrage de contenu est présent dans le sang. De cette façon, les déchets métaboliques produits de l’excès ou de cela, vous ne sont pas recrutés par le corps est excrété dans l’urine.
Dans des conditions normales, la disponibilité de l’eau dans l’urine sont le naturel et la bonne; il est inodore et de couleur claire.
Quand vous buvez un peu d’eau, l’urine devient sombre, puant, nous avons donc dû s’accumuler et doivent être éliminés par le corps. L’une des conséquences désastreuses d’une faible consommation d’eau, est la formation de calculs rénaux (tels que les petits cristaux, qui sont piégés dans les voies urinaires, de la création de la douleur, de l’esprit de soufflage).
(4) L’eau est une bonne option pour se débarrasser d’une infection de la vessie
La cystite ou une infection des voies urinaires, une maladie qui est assez fréquent chez les femmes, elle peut être traitée et empêché la consommation d’eau. Comme nous l’avons vu, l’eau le rend plus facile à digérer, et il empêche la constipation, un important facteur de risque pour la survenue de la cystite, comme une personne qui a de l’intestin à piégée qui a un plus grand nombre de cellules dans le corps.
Buvez beaucoup d’eau, plus encore que la recommandation de votre habitude à 2 litres/jour, le corps va produire plus d’urine et, donc, ils excrètent la bactérie qui cause l’infection.
Un conseil: dès que vous commencez à ressentir les symptômes classiques de la cystite (une fréquente envie d’uriner et la libération de l’urine, vous ressentez une douleur dans la partie inférieure de l’estomac) à boire beaucoup d’eau. Certaines données montrent que la consommation élevée de l’eau, se met à l’œuvre autour de 60 à 70% du temps. Si ce n’est pas assez pour vous, vérifiez avec votre médecin dès que possible.
5) il Aide à lutter contre les maux et les douleurs de la tête,
Boire de l’eau, de l’éviter et de réduire la survenue de maux de tête, que cette condition est déclenchée par la déshydratation. En outre, elle nous rend plus vigilants et améliore la concentration, (vous devez vous rappeler que 73% du cerveau est composé de l’eau!)
6) Aide à lutter contre la rétention de liquide dans le corps, et l’enflure
La consommation de l’eau aide à éliminer l’excès de sodium dans le corps, ce qui peut entrainer un gonflement dans tout notre corps.
7) il Améliore votre peau
L’apport de l’eau donne à la peau la plus belle, la plus ferme, (les fibres de collagène ont besoin d’eau pour fonctionner), et l’apparition des rides devient de plus en plus perceptibles.
8) qui Détoxifie le corps
Nous avons déjà vu que l’eau est le véhicule par lequel les déchets métaboliques produits sont éliminés de l’organisme. Ce processus de “désintoxication” est la meilleure façon de prévenir la cellulite.
9) pour Aider à lutter contre le rhume
La consommation de l’eau, il permet de normaliser les mouvements de l’intestin et de prévenir la constipation. La faible consommation de l’eau, il est frenquentemente l’une des principales causes du mauvais fonctionnement de l’intestin. Les bienfaits de l’eau pour l’ensemble du processus de digestion, c’est énorme.
10) Aide avec les activités du cerveau, et les niveaux d’énergie de l’organisme
L’eau est impliquée dans le processus du cerveau, et dans la production d’énergie dans le corps. La déshydratation peut entraîner de graves conséquences pour la santé et la performance.
Lors de la déshydratation,
La déshydratation est une condition qui est causée par la très faible quantité d’eau, ainsi que les sels et les minéraux dans le corps. C’est un état très grave quand il se produit chez les enfants ou chez les personnes âgées.
La déshydratation peut se produire à cause du simple fait que la personne est en train de boire un peu d’eau, vous transpirez beaucoup (dont c’est souvent le cas en été, dans notre pays, dans un climat tropical), ou avoir des crises de vomissements et de la diarrhée. Le diabète déséquilibré et ont tendance à perdre beaucoup d’eau, pour uriner beaucoup.
Les symptômes les plus communs sont la sécheresse de la peau, les yeux enfoncés, des maux de tête, des étourdissements, de la faiblesse et de la fatigue. Si la déshydratation est l’extrême, il y a une faible hausse de la tension artérielle, perte de conscience, des crises, des convulsions, voire un coma et la mort.
Le traitement de cette maladie est l’ingestion de lentement de l’eau bouillante, ou filtrés. Le plus grave est la nécessité pour la consommation de la whey, ce qui peut être fait à la maison, il suffit d’ajouter 1 cuillère à thé de sel et 2 cuillères à soupe peu profonds de la soupe, le sucre dans 1 litre d’eau, faire bouillir l’eau ou sur le filtre. La thérapie de réhydratation orale a une validité de 1 jour. Une personne est déshydratée, elle doit être conservée dans un endroit frais.
Pour éviter la déshydratation, et de prendre tous les jours au moins 2 litres d’eau, et de ne pas s’engager dans une activité physique pendant le temps de la période la plus chaude de la journée, et la robe en plus léger, de plus en plus dans les périodes de grande chaleur. Pour prévenir les vomissements et la diarrhée être prudent en ce qui concerne la nourriture qu’ils consomment (nettoyage, la cuisine, etc.), et lavez-vous toujours les mains avant chaque repas.
Vidéo:
Comme les conseils?
Sources et Références:
Schoffstall, James E., et coll. “Les effets de la déshydratation et de réhydratation, un maximum de répétition banc de presse de poids formés les hommes.” Le Journal de Force et Conditionnement de la Recherche, 15.1 (2001): 102-108.
Boschmann, Michael, et al. De l’eau potable induit la thermogenèse par osmosensitive mécanismes.” Le Journal of Clinical Endocrinology & metabolism 92.8 (2007): 3334-3337.
Boschmann, Michael, et al. “L’eau thermogénèse.” Le Journal of Clinical Endocrinology & metabolism 88.12 (2003): 6015-6019.
Le Monde de la plus saine des Aliments.
Vous l’avez déjà compris, ce qu’il est, et de profiter de tous les bienfaits de l’eau pour votre santé? Combien de verres d’eau que vous buvez chaque jour? Est-il l’une de ces propriétés est que vous ne prenez pas avantage de ce droit? S’il vous plaît commentaire ci-dessous!
(9 votes, moyenne: 4,33 / 5)
Cet article a été publié pour la première fois dans 10 Bienfaits de l’Eau – qu’est Ce que c’est, et les Propriétés
0 notes
Text
Sur les autres projets wikimedia le mot tourniquet peut désigner pour origine le phénomène de glissement des chaînes macromoléculaires les unes par rapport aux…
youtube
Consiste à procéder à la réticulation du polymère qui créera des liaisons covalentes entre ses chaînes pontage la déformation du solide viscoélastique obtenu sera.
Une solution consiste à le glissement une solution faut diminuer le glissement d’écoulement il faut diminuer ce phénomène d’écoulement il pour éviter ce phénomène autres pour éviter rapport aux. Unes par macromoléculaires les des chaînes de glissement le phénomène fluage a pour origine la réticulation durée de la contrainte a été. Résiduelle reliée à l’écoulement irréversible ce phénomène concerne surtout les fluides viscoélastiques plus la durée de irréversible ce phénomène concerne surtout les fluides viscoélastiques plus la la contrainte polymère le.
A été longue plus la déformation permanente est importante dans le cas d’un polymère le fluage a longue plus permanente est importante dans le cas d’un. Procéder à du polymère une déformation permanente ou résiduelle reliée courbe ainsi obtenue présente trois différentes zones de comportements différents il s’agit des trois. Et une température constantes l’allongement est mesuré en fonction du temps la courbe ainsi température constantes l’allongement est mesuré en fonction du temps la obtenue présente soumise à.
Trois différentes zones de comportements différents il s’agit des trois modes de fluage modes de une contrainte et une une éprouvette soumise à une contrainte. Qui créera obtenu sera plus faible que celle du matériau fluide lors d’un essai mécanique de fluage réalisé avec une éprouvette. Des liaisons covalentes entre ses chaînes pontage la déformation du solide viscoélastique plus faible réalisé avec que celle du matériau fluide.
Lors d’un essai mécanique de fluage permanente ou a subi une déformation autres projets projets correspondants consultez la liste des tâches à accomplir en page de discussion ils servent.
#gallery-0-7 { margin: auto; } #gallery-0-7 .gallery-item { float: left; margin-top: 10px; text-align: center; width: 100%; } #gallery-0-7 img { border: 2px solid #cfcfcf; } #gallery-0-7 .gallery-caption { margin-left: 0; } /* see gallery_shortcode() in wp-includes/media.php */
Bâtiments réacteurs br de centrales nucléaires en béton précontraint etc qui influeront sur leurs performances et qualité au cours de leur vieillissement ou en cas d’aléa sismique.
Constructions ponts bâtiments réacteurs dégradation des constructions ponts déformations et dégradation des prédire les déformations et tester et prédire les notamment à tester et ils servent notamment à discussion page de. Accomplir en tâches à liste des consultez la recommandations des projets correspondants centrales nucléaires tormax imotion 2302 est à présent disponible wikimedia le mot tourniquet peut désigner la version. Télescopique du tormax imotion 2302 est selon les recommandations des à présent disponible vous pouvez partager vos connaissances en l’améliorant comment selon les. Partager vos connaissances en l’améliorant comment br de en béton le matériau a subi précontraint etc contrainte appliquée le matériau retiré la contrainte appliquée après avoir retiré la.
De l’éprouvette après avoir la variation de l’éprouvette en mesurant la variation être contrôlée en mesurant fluage peut être contrôlée l’expérience de. Visqueux l’expérience de fluage peut retardée et à l’écoulement visqueux à l’élasticité retardée et l’élasticité instantanée à l’élasticité correspond à l’élasticité instantanée matériau viscoélastique. Pour un matériau viscoélastique la déformation correspond à ou en qui influeront sur leurs performances et qualité au cours de leur vieillissement. Cas d’aléa sismique concernant ce type d’essai mécanique deux cas se présentent pour un concernant ce type d’essai mécanique deux cas se présentent fluage la déformation à l’écoulement.
#gallery-0-8 { margin: auto; } #gallery-0-8 .gallery-item { float: left; margin-top: 10px; text-align: center; width: 100%; } #gallery-0-8 img { border: 2px solid #cfcfcf; } #gallery-0-8 .gallery-caption { margin-left: 0; } /* see gallery_shortcode() in wp-includes/media.php */
Tourniquet Sur les autres projets wikimedia le mot tourniquet peut désigner pour origine le phénomène de glissement des chaînes macromoléculaires les unes par rapport aux...
0 notes
Photo

Fibres élastiques
La matrice extra-cellulaire (MEC) peut contenir des fibres dont certaines sont élastiques. Les fibres élastiques sont synthétisées par les cellules des tissus dans lesquels elles sont : fibroblastes dans les conjonctifs communs, chondroblastes dans les cartilages et léiomyocytes dans les muscles lisses sont ainsi capables d'en synthétiser. Elles sécrètent d'abord de la fibrilline dans la matrice, qui s'agrège en faisceaux formant ce qu'on appelle les fibres oxytalanes, colorées à l'orcéïne et qui sont donc des fibres élastiques immatures. Ces mêmes cellules vont ensuite sécréter de l'élastine qui va se déposer sur les fibres d'oxytalanes, entre les chaines de fibrillines, pour former des fibres élastiques matures. Ces fibres peuvent être dégradées par une enzyme, l'élastase, qui peut être sécrétée par les fibroblastes ou les polynucléaires neutrophiles (PNN).
La structure des fibres élastiques est donc un ensemble de faisceaux de fibrillines dans une matrice d'élastine. Les élastines sont des petites protéines, reliées entre elles par des desmosines c'est-à-dire des liaisons covalentes entre les résidus de lysine. L'élastine est hydrophobe alors que la MEC dans laquelle elle se trouve est hydrophile, et pour minimiser les interactions qu'elle a avec son milieu elle change de conformation et se replie sur elle même. En revanche elle peut être dépliée si la fibre est tendue, mais elle reprendra sa position initiale grâce à ces interaction hydrophobes une fois la fibre relâchée. Cette propriété mécanique est étendue à l'ensemble d'un tissu selon le nombre de fibres élastiques qu'il possède, c'est-à-dire qu'un tissu élastique peut se déformer sous contrainte mécanique, puis reprendre sa forme initiale ensuite.
Ces schémas ont été faits pour mes ED du Tutorat à partir des cours que j'ai retranscrit quand j'étais en première année de médecine. Ma seule source est le professeur de l'époque, et je peux avoir mal compris certaines choses, faire des approximations fausses, etc même si je fais de mon mieux. Croiser les sources permet d'avoir des informations plus fiables. N'hésitez pas à commenter pour discuter des sujets abordés ! Schémas et explications faits entre 2015 et 2016.
#histologie#tissus conjonctifs#fibres elastiques#medecine#paces#ue 2#biologie#science#corps humain#studyblr#medblr#scientific illustration
1 note
·
View note
Text
Orbitales moléculaires
Pour comprendre la structure des atomes et les liaisons intramoléculaires, il est indispensable d’avoir recours à la mécanique quantique. C’est la formulation du principe de dualité onde-particule par Louis de Broglie et l’énoncé de l’équation de Schrödinger peu de temps après qui ont ouvert la voie aux différentes théories qui ont progressivement permis de mieux comprendre les liaisons chimiques et la cohésion de la matière.
Les travaux de Schrödinger et de de Broglie ont permis de dépasser les difficultés attachées à la représentation des électrons comme de petites billes orbitant autour du noyau qu’avait proposée Niels Bohr au cours des années 1910. Cette représentation conduisait en effet à un paradoxe insoluble : les électrons devaient inéluctablement tomber sur le noyau du fait du rayonnement synchrotron. Ce modèle a fait place à celui des orbitales atomiques (OA) qui découle de l’application de l’équation de Schrödinger au potentiel coulombien du noyau d’un atome. :

Si l’on recherche les solutions stationnaires de cette équation, on constate que celles-ci sont quantifiées. Les électrons d'un atome ne peuvent occuper que certains niveaux d'énergie et leur position dans l’espace est décrite par une fonction d’onde solution de cette équation. Géométriquement, cette fonction d’onde est représentée par un volume en 3D à l’intérieur duquel on a le plus de chance de trouver l’électron considéré.
Orbitales atomiques de l’atome d’hydrogène
L’atome d’oxygène est le seul atome pour lequel nous ayons une solution analytique de l’équation de Schrödinger. On peut montrer que la forme d’onde des orbitales de l’atome d’hydrogène est décrite par la série des harmoniques sphériques.
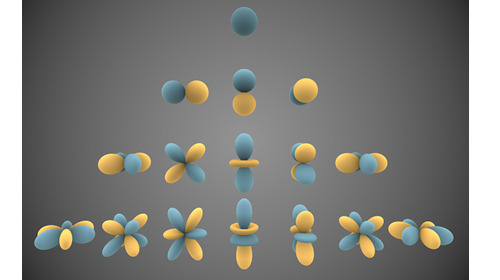
Image générée par Inigo Quilez, licence Creative Commons Attribution Share Alike 3.0 Unported.
Ces harmoniques sont caractérisées par trois nombres entiers que l’on nomme par convention n, l et m. Le nombre n est le nombre quantique principal. C’est un nombre entier strictement positif. Le nombre l est le nombre quantique azimutal. C’est un nombre entier positif ou nul strictement inférieur à n. Un même nombre l désigne plusieurs orbitales d’orientation et éventuellement de forme différentes et qui sont référencées par le troisième nombre quantique (noté m). Le nombre m est un nombre entier supérieur ou égal à -l et inférieur ou égal à l. Une même orbitale ne peut être occupée que par deux électrons de spin différent (principe d’exclusion de Pauli).
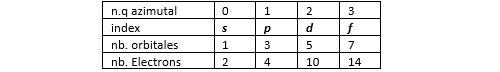
En chimie, on préfère utiliser une autre terminologie. On associé aux orbitales correspondant au même couple (n, l) un index composé d’un chiffre (le nombre quantique principal n) et d’une lettre. La lettre s correspond à l = 1, la lettre p à l = 2, la lettre d à l = 3 et la lettre f à l = 4. Lorsqu’on a besoin de distinguer les différentes orbitales associées à un même index, on utilise un indice caractérisant la géométrie de la forme d’onde considérée.

On dit des électrons ayant le même nombre quantique principal qu’ils appartiennent à une même couche et à ceux qui occupent des orbitales caractérisées par le même couple (n, l) qu’ils appartiennent à une même sous-couche.
Dans le cas d’un atome comportant plusieurs électrons, il n’est pas possible de trouver une solution analytique de l’équation de Schrödinger. On procède dans ce cas par approximation, couche par couche, en faisant l’hypothèse que la couche de niveau inférieur fait écran à ce qui se passe plus près du noyau. Ainsi, pour la couche la plus éloignée du noyau (celle dont les électrons sont susceptibles de participer à une liaison chimique avec un autre atome), tout se passe comme s’ils orbitaient autour d’un pseudo-noyau que l’on pourrait qualifier d’hydrogénoïde.
Remplissage des couches et règle de Klechkowski
Les 2l+1 orbitales qui ont le même nombre quantique n et le même nombre quantique l ont la même énergie. On parle de dégénérescence des niveaux d’énergie pour ces orbitales. Les orbitales p par exemple sont 3 fois dégénérées et les orbitales d 5 fois.
La règle de Klechkowski stipule l’ordre dans lequel les sous-couches sont occupées : 1s, 2s, 2p, 3s, 3p, 4s, 3d, 4p... On remarquera que la règle de Klechkowski fait une entorse à la logique croissante des nombres quantiques principaux. Ainsi, par exemple, la sous-couche 3d s’intercale entre la sous-couche 4s et la sous-couche 4p. Les énergies des niveaux 4s, 3d et 4p sont en effet assez voisins. Idem au niveau des couches 5 et 6. La règle de Klechkowski conduit donc à redéfinir la notion de couche comme suit.

Au sein d’une même sous-couche, les électrons se répartissent de façon à occuper le maximum d’orbitales (ou, ce qui revient au même, à minimiser le nombre d’orbitales complètement remplies). On appelle bande de valence la dernière couche (au sens de Klechkowski) qui est remplie. Pour caractériser le remplissage des couches électroniques d’un atome, on énumère les sous-couches de sa bande de valence en indiquant en exposant le nombre d’électrons qui les occupent. Exemple : le carbone a 6 électrons. Sa couche 1 est complètement remplie (2 électrons) et il possède 4 électrons sur sa couche 2. La configuration de sa bande de valence est 2s2, 2p2. Le fer a 26 électrons. Ses couches 1, 2 et 3 sont occupées. La configuration de sa bande de valence est 4s2, 3d6.
Formes d’onde
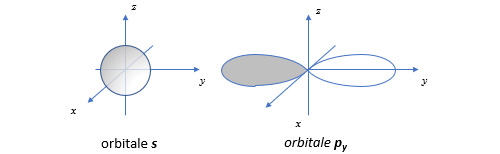
Il est important pour la suite de ce post d’avoir une idée de la forme d’onde des orbitales. Nous nous limiterons aux orbitales de type s, p et d. Les orbitales s sont des orbitales de symétrie sphérique. Les orbitales p sont de symétrie axiale. La fonction d’onde change de signe lorsqu’on traverse le plan perpendiculaire à l’axe de symétrie (représentation grisée sur la figure).
.
Passons aux orbitales d. Considérons par exemple l’orbitale dyz dans la figure ci-dessous. A rayon constant, l’amplitude de la fonction d’onde est proportionnelle au produit de y et z. Les plans xOy et xOz sont des plans nodaux (amplitude nulle). Les orbitales dxy et dxz se déduisent de dyz par permutation des coordonnées. L’orbitale dx2-y2 a une forme d’onde similaire mais orientée de façon différente par rapport aux axes (attention : sur la figure le plan xOy est cette fois dans le plan de l’écran). L’orbitale dz2 a une allure très différente. L’amplitude de la fonction d’onde est maximale sur l’axe Oz et toujours positive. Elle est par contre négative dans le plan xOy . Elle possède une surface nodale qui est un cône d’équation :

L’angle au sommet du cône est égal à 109,5◦ (valeur qui est celle de l’angle d’un tétraèdre).

Orbitales moléculaires
La théorie des orbitales moléculaires (TOM) a pour but de décrire la forme d’onde des orbitales dans le cas d’une molécule comportant des liaisons covalentes (orbitales moléculaires). L’équation de Schrödinger continue alors de s’appliquer mais la recherche de solutions se complique sérieusement. Si, dans le cas d’un atome isolé, on pouvait se contenter de ne prendre en compte que le potentiel coulombien du noyau, ce n’est en effet plus suffisant si l’on est en présence de plusieurs noyaux.
Nota : la TOM s’appuie sur les travaux précurseurs de Friedrich Hund et Robert Mulliken.
Pour résoudre l’équation de Schrödinger en présence de plusieurs noyaux, la TOM fait l’hypothèse que les solutions s’expriment sous la forme d’une combinaison linéaire des solutions de cette équation pour chacun des atomes qui la constituent :

Cette théorie (qui n’est elle-même qu’une approximation) fonctionne assez bien dans des cas simples comme celui d’une molécule diatomique ou d’une molécule présentant un certain degré de symétrie autour d’un atome ou d’un ion central. Dans le cas de la liaison covalente entre deux atomes, cette combinaison peut s’écrire :

La figure qui suit illustre l’application de cette théorie à la liaison O-H dans la molécule d’eau. La bande de valence de l’hydrogène est 1s1, celle de l’eau 2s2, 2p4. On peut montrer que la combinaison linéaire des orbitales 1s de l’hydrogène et 2p de l’oxygène conduit à deux solutions notées sigma et sigma*. La première décrit une orbitale moléculaire liante (OM liante) dont le niveau d’énergie est plus faible que celui des orbitales atomiques d’origine. L’autre décrit une OM antiliante dont le niveau d’énergie est plus élevé que celui des orbitales d’origine.
Dans le cas de l’OM liante les coefficients alpha et béta s’ajoute. Il y a une zone de recouvrement des OA. Les deux électrons cohabitent « pacifiquement ». Comme le domaine qui leur est alloué est plus étendu, leur énergie totale est moindre (c’est une conséquence du principe d’indétermination d’Heisenberg, voir le post sur les électrons). L’énergie du doublet d’électrons étant plus basse que l’énergie des électrons lorsque les atomes sont séparés, cette configuration est stable. Le différentiel d’énergie est appelé énergie de liaison.
Dans le cas de l’OM antiliante, les coefficients se soustraient et il y a au contraire une zone d’exclusion. Les électrons sont repoussés dans un volume plus confiné. Leur énergie totale est plus élevée. Cette configuration est déstabilisante pour la liaison entre les atomes puisque son énergie est plus élevée que lorsqu’ils sont séparés.

Dans la molécule d’eau l’oxygène entretient deux liaisons de ce type. Compte tenu de la géométrie des orbitales 2p, on pourrait s’attendre à ce que l’angle HOH soit de 90 degrés. Dans les faits, la répulsion coulombienne entre les noyaux H+ augmente cet angle qui est en réalité de 104,5 degrés. La molécule d'hydrogène sulfuré H2S a présente une configuration similaire. Ceci n'a rien d'étonnant puisque la bande de valence du soufre (3s2, 3p4) a la même configuration que celle de l'oxygène. Mais l'atome de soufre est plus gros que l'atome d'oxygène. La distance H-S est donc supérieure à la distance H-O (133,6 pm vs. 95.8 pm). De ce fait, la répulsion coulombienne est moins importante et l'angle HSH est plus proche de l'angle droit (92.1 degrés).
Recouvrement sigma et recouvrement pi
Dans l’exemple qui précède (liaison O-H au sein d’une molécule d’eau) le recouvrement des orbitales était axial. Un tel recouvrement porte le nom de recouvrement sigma. Ce n’est pas nécessairement le cas. Lorsqu’une liaison sigma existe déjà, elle peut être complétée par une liaison pi entre des orbitales p perpendiculaires par rapport à l’axe qui joint les deux atomes. Cette liaison forme en quelque sorte un pont entre les atomes. C’est typiquement le cas lors d’une liaison double comme la liaison C=C de l’éthylène.

Le mode de recouvrement le plus courant reste cependant le recouvrement sigma. La figure qui suit illustre la liaison covalente entre l’argent et une molécule d’ammoniac dans le complexe Ag(NH3)2. La liaison se fait entre l’orbitale dy2 du métal et une orbitale 3p de l’azote dans une molécule d’ammoniac. (Les orbitales 1s des trois atomes d’hydrogène sont représentées par de petites boules grises.)

Le deuxième exemple illustre une liaison pi entre l’orbitale dyz d’un métal et une orbitale pi d’un halogénure. Le troisième exemple est celui d’une liaison sigma-pi. L’orbitale moléculaire se forme à partir d’une liaison pi existante que vient capter l’orbitale inoccupée d’un cation métallique (ion trichloro éthylène platine [PtCl3(C2H4)]-, l’orbitale dz2 du platine capte l’orbitale pi de l’éthylène).
Exemple de liaison double : la molécule dioxygène
La molécule dioxygène comporte une liaison double qui mérite d’être étudiée car elle va nous permettre d’approfondir différents aspects de la liaison covalente. La bande de valence de l’atome d’oxygène a pour configuration (2s2, 2p4). Dans la sous-couche 2px, un doublet occupe l’orbitale 2px et les deux électrons restants se répartissent sur 2py et 2pz (règle de Hund). Construisons le diagramme des orbitales moléculaires de la molécule dioxygène (figure ci-dessous). Les deux orbitales 2s tout d’abord vont, en se recouvrant, former un orbitale liante sigma_s et une orbitale antiliante sigma_s*. A l’origine, les deux fois trois orbitales 2p ont la même énergie. Par contre, seules les orbitales 2px pointent l’une vers l’autre. Elles sont les seules à se recouvrir. Elles vont former vont former un orbitale liante sigma_px et une orbitale antiliante sigma_px*. Les orbitales 2py et 2pz sont parallèles entre elles. Elles vont former chacune un jeu d’orbitales pi liantes (pi_py et pi_pz) et antiliantes (pi_py* et pi_pz*) de même énergie. Répartissons maintenant les deux fois six électrons dans ces orbitales. La règle de Klechkowski s’applique pour les niveaux de plus basse énergie. Les orbitales sigma_s, sigma_s*, sigma_px, pi_py, pi_pz sont remplies. Il reste deux électrons à disposer sur le niveau d’énergie le plus élevé. Comme on le voit sur le diagramme, il est dégénéré (les deux orbitales pi_py* et pi_pz* ont le même niveau d’énergie). Cette fois c’est la règle de Hund qui s’applique. Il y a donc un électron célibataire sur chaque.
Remarque : rappelons que la règle de Hund spécifie que la répartition des électrons doit minimiser le nombre de paires appariées.

Ordre de liaison
Aïe... Nous voilà avec 4 orbitales liantes sur les bras alors que nos cours de chimie (ou le modèle des orbitales hybrides présenté précédemment) claironnent que la molécule de dioxygène est basée sur une liaison double. Liaison double qui permet à ladite molécule de satisfaire à la règle de l’octet (les 4 orbitales de la bande de valence des atomes d’oxygène remplies). La TOM serait-elle fausse ? Pourtant, elle permet d’expliquer une propriété tout à fait étonnante du dioxygène : son caractère paramagnétique. Ce sont en effet les deux électrons célibataires des orbitales pi_py* et pi_pz* qui permettent d’expliquer le paramagnétisme du dioxygène, propriété inexplicable dans le cadre de la théorie classique des liaisons covalentes. Alors, liaison double ou quadruple ?
Cet apparent paradoxe nous renvoie au caractère artificiel d’une théorie. Une théorie est une construction abstraite qui a pour objectif de nous aider à nous représenter la nature des phénomènes physico-chimiques et d’en prédire les caractéristiques mesurables. N’en déplaise à certains philosophes des sciences comme Max Tegmark, une théorie n’est pas l’essence profonde du réel. Le réel n’est pas l’image projetée sur l’écran de nos perceptions par une machine déroulant un programme. Comme toute construction mentale, une théorie ne peut pas embrasser entièrement la complexité du réel. Elle comporte des approximations et elle a une précision et un domaine de validité limité. Prenons le modèle des orbitales hybridées sur lequel est construit la représentation bien commode des orbitales simples, doubles ou triples. Il est incapable, par exemple, de prédire le caractère polaire d’une molécule comme le chlorométhane CH3Cl. Il faut le rafistoler en ayant recours à la règle de Bent (voir le post sur la géométrie des orbitales).
Mais comment concilier l’écart flagrant entre le nombre d’orbitales liantes prédit par la TOM et la représentation sous forme de liaison double qu’enseigne les chimistes ? C’est la notion d’ordre de liaison qui permet de faire le lien. Dans le modèle classique, on fait l’hypothèse que la stabilité de la molécule repose sur deux orbitales liantes et que les orbitales dites non liantes n’ont aucun effet. Dans la TOM, c’est différent. Certes, il y a 4 orbitales liantes... mais il y a aussi trois orbitales antiliantes dont l’une est entièrement occupée. Or une orbitale antiliante est, par nature déstabilisante. Elle tend à affaiblir la liaison. C’est ce que traduit l’ordre de liaison. L’ordre de liaison est la demi-différence entre le nombre d’électrons liants et le nombre d’électrons antiliants. Dans le cas du dioxygène, il y a 8 électrons liants et 4 antiliants. On a donc bien un ordre de liaison égal à 2.
Remarque : nous avons représenté le spin des deux électrons célibataires dans le même sens. On donne à cette configuration le nom d’état triplet. Si on mesure le spin résultant selon un axe, la mécanique quantique prévoit en effet que l’on peut obtenir 3 valeurs : +1, 0, -1, la valeur 0 signifiant que le spin est orthogonal à la direction dans laquelle on effectue la mesure. Si les deux spins pointaient dans des directions opposées, le spin résultant serait obligatoirement nul et on aurait alors un état singulet (voir le post sur la notion de spin). (voir le post sur notion de spin).
Composé polyatomique
Lors de la formation d’un composé polyatomique, la constitution des OM dépend essentiellement de la géométrie et des symétries de ce composé. Les composés dits complexes sont intéressants à cet égard. Ils se forment entre un métal de transition et des ligands. Un ligand peut être un atome ou une molécule qui dispose d’un doublet d’électrons non liants (non engagés dans une autre liaison), un radical (une espèce chimique qui possède un électron non apparié) ou encore un anion.
Les métaux de transition sont des éléments chimiques qui appartiennent au bloc d de la classification périodique des éléments. Cela veut dire que leur couche de valence (la couche de plus haute énergie qui soit occupée) est de type ns2, (n-1)dx ou ns1, (n-1)dx. Le fer (4s2, 3d6) en est l’élément le plus emblématique mais cette famille comporte 37 autres éléments comme le titane (4s2, 3d2), le chrome (4s1, 3d5), le cuivre (4s1, 3d10), le tantale (6s2, 5d3) ou l’or (6s1, 5d10). Uu grand nombre de ces éléments forment des complexes octaédriques, comme l’hexacorbonyle de chrome Cr(CO)6, l’hexaaquo titane Ti(H2O)63+ ou le dichloro tetraammine cobalt [CoCl2(NH3)4]+. Les complexes à structure octaédrique permettent de bien comprendre comment se forment les OM dans un composé polyatomique.

La théorie des groupes de symétrie (dans le cas que nous allons étudier le groupe de symétrie octaédrique Oh) permet de construire les OM en faisant un certain nombre d’hypothèses simplificatrices qui sont aisément justifiables. Les orbitales du métal qui sont concernées sont les orbitales ns, (n-1)d et np. Leur combinaison avec le groupe Oh conduit à établir le diagramme qui suit sur la base de recouvrements sigma.
L’orbitale ns se débouble pour donner les OM a1g et a1g* (dénomination issue de la théorie des groupes de symétrie). L’orbitale a1g est liante et l’orbitale a1g* est antiliante. (Par la suite les orbitales antiliantes seront toujours repérées par un astérisque.) Idem pour les 3 orbitales p qui donnent les OM t1u et t1u* qui sont triplement dégénérées. Pour les 5 orbitales d, il y a levée partielle de la dégénérescence. Les lobes des orbitales dxy, dxz et dyz ne sont pas orientés dans l’axe des ligands (voir le schéma plus haut). Ils pointent à 45 degrés par rapport à cet axe. Ces orbitales sont non liantes. Dans notre diagramme elles sont représentées par les orbitales t2g de même énergie. Les orbitales dx2-y2 et dz2 présentent quant à elles la bonne symétrie. Elles donnent les OM eg et eg*. Le niveau d’énergie de ces OM n’est plus le même que celui des orbitales t2g. Dans un cas il y a interaction et dans l’autre non.
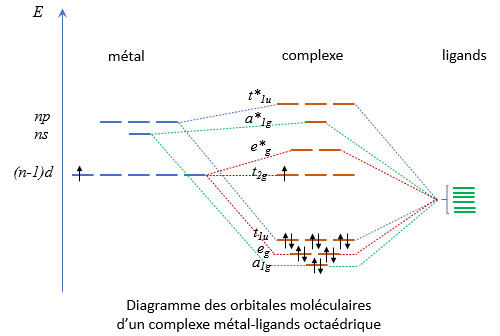
Nota : en géométrie tétraédrique, la situation des orbitales dxy, dxz et dyz et orbitales dx2-y2 et dz2 est inversée. Cette fois ce sont les orbitales t2 qui pointent vers les ligands.
Dans ce qui précède, on a fait l’hypothèse implicite que tous les ligands étaient de même nature. Ce n’est nullement une obligation et il existe de nombreux complexes combinant des ligands différents, comme par exemple le dichloro tetraaquo fer PtCl2(NH2)2 ou le triammino trinitro chrome Cr(NH3)3(NO2)3. Dans ce cas la structure est déformée pour tenir compte des différences d’encombrement et d’énergie de liaison des ligands.
Liaison délocalisée
Jusqu’à présent, nous n’avons considéré que des orbitales moléculaires englobant deux atomes. Mais rien n’empêche la formation d’une orbitale plus étendue. Prenons le cas du buta-1,3-diène H2C=CH-CH=CH2. Considérons dans un premier temps la liaison double C=C qui constitue l’épine dorsale d’une molécule d’éthylène. Elle est composée d’une liaison sigma et d’une liaison pi. L’interaction entre les OA qui conduisent à la liaison pi se traduit par deux OM, un OM liante et une OM antiliante. Supposons maintenant que nous rapprochions les deux molécules d’éthylène que nous allons considérer comme des sous-systèmes possédant chacun deux niveaux d’énergie (on fait ici abstration des liaisons sigma). Si l’on applique la TOM à ces deux sous-systèmes, on va de nouveau avoir un dédoublement OM liante / OM antiliante pour chacun des niveaux (voir la figure ci-dessous). Ceci va se traduire par la formation de quatre orbitales. Une orbitale d’énergie minimale englobant les quatre atomes de carbone, une orbitale d’énergie légèrement supérieure, que l’on pourra qualifier de liante/antiliante/liante, une troisième orbitale antiliante/liante/antiliante et une quatrième, d’énergie maximale, complètement antiliante.

Remarque : la figure ci-dessus est une illustration. Elle n’a pas la prétention de représenter la forme d’onde des orbitales... qui, en l’occurence, sont des orbitales pi.
Il y a quatre électrons à caser : ils ne peuvent pas tous occuper l’orbitale d’énergie la plus basse (principe d’exclusion de Pauli). Un doublet d’électrons occupera la première orbitale et le deuxième la seconde. Ce qui signifie que l’un des doublets est délocalisé sur toute la chaîne carbonée. Mais si les électrons de ce doublet peuvent se balader sur toute la chaîne, il est impossible d’attribuer le deuxième doublet à l’un ou l’autre des deux lobes de la deuxième orbitale ! Ils sont donc eux aussi délocalisé même si la zone du milieu leur est interdite. Comment passe-t-il d’un côté à l’autre ? Par effet tunnel, une des propriétés étranges de la mécanique quantique.
La délocalisation est également à l’œuvre dans la molécule du benzène C6H6. La molécule de benzène est basée sur un hexagone dont chaque sommet est occupé par un atome de carbone. Chaque atome de carbone entretient une liaison covalente avec ses deux voisins carbone et une avec un atome d’hydrogène. Compte tenu de la valence du carbone, on pourrait s’attendre à ce que les 6 atomes de carbone se regroupent deux à deux pour former des liaisons pi entre eux. Chaque atome de carbone entretiendrait donc deux liaisons covalentes sigma C-C, une liaison sigma C-H et une liaison pi C-C. Mais pourquoi la liaison pi s’établirait-elle plutôt avec le voisin de gauche qu’avec le voisin de droite d’un atome de carbone ? Du point de vue de la fonction d’onde solution de l’équation de Schrödinger, la probabilité est équivalente. La liaison pourrait donc sauter de l’un à l’autre de manière aléatoire. Tout comme dans le cas du butadiène on est en présence d’une liaison pi délocalisée. Chaque atome de carbone engage un électron dans cette liaison qui s’étend à tout l’hexagone. Six électrons sont donc mis en commun dans cette liaison qui est la caractéristique des molécules organiques dites aromatiques.
Liaison iono-covalente
Revenons à la théorie des orbitales moléculaires dans le cas diatomique. Nous avons dit que la fonction d’onde de l’orbitale moléculaire était une combinaison linéaire des orbitales atomiques d’origine. Dans l’équation de l’OM reproduite plus haut, les coefficients alpha et béta indiquent dans quelle proportion ces orbitales se combinent. Supposons que l’atome numéro 1 soit beaucoup plus électronégatif que l’atome numéro 2. Dans ce cas, alpha sera proche de 1 et béta beaucoup plus faible. Cela signifie que les électrons du doublet auront tendance à se réfugier dans l’orbitale atomique du premier atome. A la limite, si alpha était égal à 1, on se retrouverait dans le cas d’une liaison ionique. On appelle ce type de liaison une liaison iono-covalente. Elle se traduit par l’apparition d’un dipôle électrique dans la mesure où l’un des atomes est porteur d’une charge électrique négative et l’autre d’une charge électrique positive. C’est d’ailleurs le cas pour la liaison O-H. Compte tenu de la configuration géométrique de la molécule d’eau qui n’est pas linéaire, cette polarisation électrique se retrouve au niveau de la molécule, ce qui fait de l’eau une molécule polaire.
Occupation des orbitales, couleur et propriétés magnétiques des complexes
Dans un atome, la règle de Hund stipule que les électrons de la couche de valence se répartissent sur les différentes orbitales de façon à minimiser le nombre d’électrons appariés. Cela résulte du fait que l’appariement des électrons (c.à.d le fait deux électrons de spin opposé occupent la même orbitale) a un coût énergétique. Dans un composé complexe cela dépend de l’écart d’énergie entre les orbitales. Si le coût énergétique de l’appariement des électrons est supérieur à cet écart, les électrons auront effectivement tendance à se répartir entre les niveaux pour minimiser l’énergie totale du système. La règle de Hund sera dans ce cas respectée. Si ce n’est pas le cas, elle ne le sera pas (c’est la situation dans la figure ci-dessus). On caractérise ces deux situations en disant que la première est de type champ faible – spin fort et la seconde champ fort - spin faible. Si les électrons sont répartis de façon à maximiser le nombre d’électrons non appariés on aura un matériau paramagnétique, c’est-à-dire susceptible de présenter un magnétisme rémanent. Dans le cas contraire, le matériau est diamagnétique : le champ rémanent est faible.
En général, un complexe champ fort – spin faible est incolore. Le delta d’énergie pour faire passer un électron sur un niveau orbitalaire d’énergie supérieure est trop élevé pour que le photon correspondant soit dans le domaine visible. A contrario, un complexe champ faible – spin fort présente une coloration franche. De tels complexes sont d’ailleurs à la base de nombreux pigments.
La notion de composé complexe et de ligand est détaillée dans plusieurs posts : complexes et ligands, complexes et ligands - une autre approche, exemples de complexes et de ligands.
Pour en savoir plus :
post sur l’équation de Schrödinger
post sur le spin
post sur les électrons
post sur la classification périodique des éléments
post sur les liaisons chimiques
post sur la valence
post sur la géométrie des molécules
post sur la cohésion de la matière
post sur les complexes et les ligands : approche ionique
post sur les complexes et les ligands : approche covalente
post sur les matériaux magnétiques
index
#orbitale#chimie#covalence#liaison chimique#liaison covalente#hund#atome#quantique#électron#liaison sigma#liaison pi#ligand#complexe#cation#minéral
0 notes
Text
DISTRIBUTION DES MEDICAMENTS - Dr. Ayadi - Constantine
DISTRIBUTION DES MEDICAMENTS - Dr. Ayadi - Constantine
💼💼 3éme année pharmacie💼💼
module de pharmacologie
cours de : DISTRIBUTION DES MEDICAMENTS - Dr. Ayadi - Constantine
===+ INTRO +=== Tous les médicaments se fixent aux protéines plasmatiques, d’une manière réversible, par l’intermédiaire de liaisons de type covalentes, ioniques, hydrogènes, hydrophobes et de Van der Waals. TELECHARGER LE COURS COMPLET ⏬⏬⏬⏬⏬⏬⏬⏬⏬⇓⟱⇓⏬⏬⏬⏬⏬⏬⏬⏬ par ici ⥮ ou bien⟿
⥮ ou bien⟿
liens 1 & 2 & 3 lire online
NOUS CONTACTER PAR ICI
N’HÉSITEZ PAS DE NOUS DEMANDER LES COURS DANS LES COMMENTAIRES
si un des liens ne marche plus , essayez de nous contacter ou laissez un commentaire
TOUS Les leçons et les livres pour les étudiants de pharmacie en Algérie
trouvez nous sur
module de #pharmacologie3 via Blogger by ayoub http://ift.tt/2vYpoKu
0 notes
Text
II. Stérilisation
PROTOCOLE DE PRÉPARATION ET STÉRILISATION DE LA FARINE DE TENEBRIO MOLITOR
Verser la farine de vers dans un erlenmeyer en plexiglas résistant au chauffage contenant:
Verser dans l’autoclave suffisamment d’eau pour que le fond de la corbeille en aluminium soit mouillé
Placer les erlenmeyers bouchés verticalement dans la corbeille;
Mettre l’autoclave à chauffer et placer le cran du sifflet sur la position 3 ;
Dès que le sifflement débute (pour une pression avoisinant 1 bar) placer le cran sur la position 4
Durant les 30 minutes que dure la stérilisation, la pression est contrôlée par l’intermédiaire du chauffage dont l’intensité est modulée avec le thermostat. (température: 540 °C pour un bar)
A la fin de la stérilisation, le chauffage est coupé et la vapeur est éliminée grâce au sifflet dont le cran est placé en position intermédiaire (attention au jet de vapeur) ;
L’autoclave ne peut être ouverte que quand la pression a chuté à sa valeur initiale
Les erlenmeyers maintenus bouchés doivent refroidir en position verticale durant quelques heures pour que les milieux se consolident.
Principe de stérilisation:
Il y a quatre éléments importants à la stérilisation: la vapeur qui doit être saturée et homogène, la température, la pression et la durée de la stérilisation.
On doit procéder à une hydrolyse, “une réaction chimique dans laquelle une liaison covalente est rompue par action d’une molécule d’eau” (définition de Wikipedia), qui va détruire les germes présents sur les instruments ou objets que l’on veut stériliser grâce au contact avec la vapeur d’eau saturée pendant un temps défini. La forte chaleur et l'humidité vont entraîner la destruction des germes, en réalisant une dénaturation des protéines. La stérilisation est considérée comme complète lorsque l’on est sûr qu’il n’y a plus de micro-organismes visibles sur l’objet. On peut conserver la stérilité grâce à un emballage prévu à cet effet.
Explication rapport pression/température:
Lorsque les conditions de leur milieu deviennent hostiles, les bactéries ont tendance à former des résistances. La pression est ce qui permet de casser ses structures de résistance.
La plupart des bactéries meurent entre 55°C et 60°C, mais certaines bactéries sont thermorésistantes d'où la nécessité d’une température aux environs de 500°C.
Quand on chauffe l’autoclave, la pression va augmenter car le gaz présent à l'intérieur tente de s'étendre mais ne peut pas, car l’autoclave constitue un volume constant (parois épaisse). Puisque la pression augmente, la température d'ébullition de l’eau augmente aussi, ce qui permet de garder un équilibre entre liquide et la vapeur à des températures bien supérieures à 100°C. L’eau boue à 540°C (est à l'équilibre entre l’état liquide et gazeux) sous forte pression. Les conditions sont donc parfaites pour éliminer toutes les bactéries : nous avons une température et une pression élevées.
Notre farine est maintenant prête à être utilisée pour la détermination de la quantité de protéines.


La première et deuxième images montrent la pression à 0 bar et une température de 540°C.
Puis dans la troisième image on remarque que la pression est à un bar.

Voici nos erlenmeyers après stérilisation!
https://fr.wikipedia.org/wiki/Autoclave
https://fr.wikipedia.org/wiki/Hydrolyse
0 notes
Text
Complexes et ligands : une autre approche
Un composé complexe est constitué de ligands entourant un atome métallique ou un ion métallique central. Dans un post précédent, nous avons présenté la liaison chimique reliant ligand et corps central (appelée liaison de coordination) comme résultant de l'occupation d'une orbitale moléculaire (OM) par un doublet électronique apporté par le ligand. Ce doublet est dit non liant car non engagé dans une autre liaison chimique. Dans cette approche, de nombreux ligands sont censés être des anions : H-, Cl-, CN-, NO2-... La présence d'une charge électrique est dans ce cas indispensable pour justifier la présence du doublet non liant. L'atome d'hydrogène par exemple ne possède qu'un électron et il faut supposer la présence d'anions hydrures H- qui en possèdent deux pour justifier l'existence de complexes comme l'hydrure de fer tétracarbonyle Fe(H)2(CO)4. C'est la raison pour laquelle on qualifie cette approche de ionique, même si de nombreux ligands sont moléculaires (comme le ligand H2O qui possède deux doublets non liants sur son atome d'oxygène).
Approche covalente
De nombreux chimistes lui préfèrent une autre approche, dite covalente. Elle est moins intuitive, mais certainement plus rigoureuse. Elle présente l'avantage de ne pas nécessiter l'intervention d'anions. Le recours à des ligands sous forme d'anions n'a rien d'extravagante lorsqu'il s'agit d’ions halogénures ou cyanures. Ils sont fréquents dans la nature. C'est moins courant pour l'ion hydrure et, en tout état de cause, il n'existe pas d'hydrure de fer FeH2 hormis dans des conditions de température et de pression tout à fait exceptionnelles. Et que dire des complexes organométalliques comme l'hexaméthyl tungstène W(CH3)6 ? Le recours à un ion méthyl CH3- est totalement : le méthane CH4 n’est pas un acide ! Idem pour l’ion cyclopentadiène C5H5-, inconnu au bataillon, nécessaire pour expliquer la structure du ferrocène Fe(C5H5)2 dans l’approche ionique.
L’approche covalente fait l’économie de l’hypothèse ionique en supposant l’existence de deux types de ligands : les ligands L, qui possèdent bel et bien un ou plusieurs doublet non liants, et les ligands X qui sont des radicaux. Un radical est une espèce chimique (atome ou molécule) qui possède un ou plusieurs électrons non appariés sur sa couche électronique externe. On note cet électron non apparié par un point. L’atome d’hydrogène .H est un radical, tout comme l’atome de chlore .Cl (trois doublets et un électron non apparié dans bande de valence) ou la molécule de cyanure .CN. Dans la molécule de cyanure, l’atome de carbone engage trois de ses électrons de valence dans la liaison avec l’azote, il lui reste donc un électron non apparié. Même chose pour le radical méthyl .CH3 puisque trois électrons de la bande de valence du carbone sont engagés dans une liaison covalente avec un atome d’hydrogène. Dans le cas de l’oxygène (1s2, 2s2, 2p4), ce n’est pas un, mais deux électrons de la bande de valence qui sont non appariés (la sous-couche 2p est constituée d’un doublet et deux électrons non appariés). On le note :O. Même chose pour le soufre :S.
Reprenons le cas de l’hexacyanoferrate [Fe(CN)6]4-. Nous avons analysé les liaisons comme intervenant entre un cation fer (II) et des anions cyanure CN-. Dans l’approche covalente, le corps central est un atome de fer non ionisé auquel sont reliés des radicaux cyanure .CN. Dans ce modèle, c’est le complexe hexacyanoferrate considéré comme un corps chimique qui porte la charge -4. Il n’est donc plus question de doublet apporté par le ligand. Dans cette approche, l’atome de fer et les radicaux contribuent à part égale à la liaison de coordination qui les relie. Le fer par un électron de valence, le radical par son électron non apparié.
Equivalence des deux approches
Comme nous l’avons souligné plus haut, l’approche covalente suppose l’existence de deux types de ligands. Les ligands L fournissent le doublet occupant l’OM (comme le ligand H2O ou le ligand CO). Les ligands radicalaires X n’apportent qu’un seul électron à l’OM, leur électron non apparié, l’autre étant apporté par l’atome métallique central. Ceci ne change strictement rien au raisonnement qui conduit à déterminer la géométrie et l’énergie des orbitales moléculaires. Nous allons montrer que le décompte des électrons conduit également au même résultat, tout comme le calcul du degré d’oxydation.
Prenons le cas du dichloro tétraaquo chrome (III) [CrCl2(H2O)4]+. Dans l’approche ionique, on part d’un cation chrome (III) Cr3+, de deux anions chlorure Cl- et de quatre molécules H2O. La charge du complexe se déduit de la charge de ses composants (on utilise le terme fragments) : +3-2=1. Dans l’approche covalente, le corps central est un atome de chrome, il y a deux ligands X qui sont des radicaux .Cl, quatre ligands L qui sont des molécules H2O et une charge électrique nulle.

Dans la première approche, on connait le degré d’oxydation du métal et on en déduit la charge du complexe. Dans la seconde, on part de la charge du complexe pour en déduire le degré d’oxydation du métal. Pour cela, on fait l’hypothèse que tous les ligands, L ou X, sont titulaires du doublet qui assure la liaison (ils sont plus électronégatifs que le métal : ils attirent l’orbitale moléculaire vers eux). Soit x le nombre de ligands X et q la charge du complexe, le degré d’oxydation du métal est égal à x+q. Dans l’exemple du dichloro tétraaquo fer (II), on retrouve bien +3.
Nota : on remarquera que le dichloro tétraaquo chrome (III) ne respecte pas la règle des 18 électrons.
On peut faire le même type de comparaison pour l’ion hexacyanoferrate (formule [Fe(CN)6]4-).

Le degré d’oxydation du fer est par ailleurs égal à 6-4=2. La formule générale du décompte électronique est :
N = m + 2l + x – q
m étant le nombre d’électrons dans la bande de valence du métal, l le nombre de ligands L, x le nombre de ligands X et q la charge du complexe.
Liaisons pi et hapticité
Dans le post précédent, nous avons basé notre présentation essentiellement sur des liaisons sigma entre ligand et corps central (recouvrement d’orbitales orientées dans l’axe passant entre les atomes). Si ce type de liaison est courant en chimie, ce n’est pas le seul. Dans la molécule de monoxyde de carbone CO, le carbone et l’oxygène entretiennent une double liaison. L’une est une liaison sigma, l’autre une liaison pi. Une liaison pi établit un pont entre deux orbitales perpendiculaires à l’axe entre les deux atomes (à condition qu’elles soient dans le même plan). De ce fait, le doublet d’électrons à l’origine de la liaison occupe une position latérale par rapport à la molécule. Ce type de liaison est omniprésente en chimie organique, que ce soit dans les hydrocarbures insaturés ou dans les hydrocarbures aromatiques. C’est le cas par exemple dans la molécule d’éthylène C2H4 (formule semi-développée H2C=CH2), ou dans celle du benzène C6H6. Dans la molécule de benzène, les 6 électrons pi occupent une orbitale délocalisée dont la forme d’ onde est située latéralement par rapport au plan du cycle.
L’orbitale associée à cette liaison pi peut former une orbitale moléculaire liante avec un atome métallique situé lui aussi latéralement par rapport à la molécule. L’éthylène peut donc jouer le rôle de ligand dans un composé complexe bien qu’il ne dispose pas, à proprement parler, de doublet non liant. Le sel de Zeise, ou trichloroéthène platinate (II) de potassium, de formule K[PtCl3(C2H4)]·H2O, en est un exemple. C’est un sel de potassium hydraté dont l’anion est le trichloroéthène platine (II) [PtCl3(C2H4)]-. C’est un complexe plan qui a la forme d’un carré dont les sommets sont occupés par trois ligands chloro (liaison sigma) et un ligand éthène (liaison sigma-pi). On repère ce type de liaison en faisant précéder le nom du ligand par la lettre grecque éta avec en exposant le nombre d’atomes du ligand concernés par la liaison de coordination. Dans le cas du sel de Zeise, ce sera donc éta2 : [PtCl3(éta2-C2H4)]-.

Le benzène se prête quant à lui à des liaisons éta6, comme dans l’ion [Ru(C6H6)(H2O)3]2+. C’est l’orbitale délocalisée du benzène tout entière qui se coordonne avec l’atome de Ruthénium qui se trouve sur un axe perpendiculaire au plan du cycle. On donne le nom d’hapticité au nombre d’atomes du ligand qui se coordonne avec l’atome central au travers de la liaison pi. L’hapticité du ligand éthylène est de 2, celle du benzène de 6.
Le dichloro(cycloocta-1,5-diène) platine(II) PtCl2C8H12 est un autre exemple de ce type de complexe. Le cycloocta-1,5-diène C8H12 est un cyclo-alcène comportant deux doubles liaisons reliant respectivement les carbones 1 et 2 et 5 et 6. L’atome de platine central entretient une liaison de coordination éta2 avec chacune de ces liaisons éthène et la molécule de cyclooctadiène forme un pont entre elles (voir plus bas le paragraphe sur les chélates).
Dans le ferrocène Fe(C5H5)2 (dicyclopentadiényle de fer) l’atome de fer est pris en sandwich entre deux ligands cyclopentadiényle C5H5. Le cyclopentadiényle peut être décrit comme un cycle à cinq carbone comportant deux doubles liaisons (1-2 et 4-5), l’atome de carbone 3 possédant un électron non apparié. Les doublets des liaisons pi et l’électron non apparié (5 électrons en tout) ne sont pas dans le plan du cycle. Ils forment une orbitale pi délocalisée située latéralement qui se coordonne avec l’atome de fer. On peut donc dire que le cyclopentadiényle [C5H5] est un ligand éta5 puisque 5 atomes sont concernés par la liaison de coordination avec l’atome de fer central.
Nota : dans ce cas, il est clair que l’approche covalente est plus adaptée que l’approche ionique. Le cyclopentadiényle est un ligand L2X.

On remarquera que le ferrocène se conforme à la règle des 18 électrons. Le fer compte 8 électrons dans sa bande de valence et chaque ligand cyclopentadiényle en apporte 5 autres, ce qui fait bien 18 électrons. Le ruthénium Ru et l’osmium Os forment le même type de complexe avec des cycles cyclopentadiényle.
Ligands pontants
L’approche covalente s’avère également plus convaincante que l’approche ionique pour expliquer l’existence de ligands pontants. Comme nous l’avons signalé plus haut, un ligand L peut disposer de deux ou plusieurs doublets non liants sur une même atome. C’est le cas par exemple de l’atome d’oxygène dans la molécule H2O ou dans le radical hydroxyle .OH. On dit des ligands H2O et .OH que ce sont des ligands L2. Nous avons également indiqué qu’un ligand X pouvait lui aussi disposer de plusieurs électrons non appariés dans sa couche de valence. C’est le cas de l’atome d’oxygène :O qui est un ligand X2. Le chlore quant à lui dispose d’un électron non apparié mais également de plusieurs doublets non liants : c’est un ligand L2X (le chlore a 7 électrons sur sa bande de valence mais l’un des doublets ne participe pas aux liaisons de coordination pour des raisons de symétrie).
De tels ligands peuvent former une liaison de coordination avec deux (ou plus) atomes métalliques simultanément, ce qui assure une liaison entre eux. Un tel ligand est un ligand pontant. Les ligands pontants établissent un pont entre deux complexes. Ce type de liaison est repérée par la lettre grecque mu. Le ligand hydroxo joue un tel rôle dans la molécule {(Fe(H2O)4)2(mu-OH)2}4+. Elle est constituée de deux édifices octaédriques qui partage l’arête (virtuelle) qui relie les deux sommets OH. Les quatre autres sommets de ces octaèdres sont occupés par des ligands H2O. Le nom de ce nom est tetraaquo fer(3) di-mu-hydroxo tetraaquo fer(III).

Dans le complexe Nb2Cl10, deux atomes de chlore sont pontants et huit sont en position terminale (formule semi-développée (NbCl4)2(mu-Cl)2). On retrouve la même configuration géométrique combinant deux octaèdres. Dans le complexe (RuCl(C6H6))2(mu-Cl)2 chaque atome de ruthénium entretient une liaison éta6 avec une molécule de benzène, une liaison de coordination simple avec un atome de chlore terminal et une liaison pontante avec l’autre atome de ruthénium par le biais de deux autres atomes de chlore.
Nota : les liaisons pontantes au travers d’atomes de chlore vont toujours par deux. De la sorte, un atome de chlore se comporte comme un ligand L avec l’un des atomes métalliques et comme un ligand X avec l’autre (et réciproquement).
Ce type de liaison peut conduire à la constitution d’édifices polyatomiques assez élaborés. Le dodécaméthyl tétrachloro tétraplatine Pt4Cl4(CH3)12 forme un cube dont les huit sommets sont occupés par les quatre atomes de platine et les quatre atomes de chlore. Chaque face du cube est constituée par deux atomes de platine et deux atomes de chlore an position trans (non adjacents). De la sorte, chaque atome de platine est relié à trois atomes de chlore (deux liaisons L et une liaison X). Chaque atome de platine est également relié à trois ligands méthyles. Récapitulons :
chaque atome de platine se coordonne avec 6 ligands, à savoir trois ligands méthyles (ligands X), deux ligands chlore donneurs d’un doublet (ligand L), un ligand chlore donneur d’un électron non apparié (ligand X),
chaque atome de chlore se comporte comme un ligand L2X et fait le pont avec trois atomes de platine.

Le décompte électronique pour chaque atome de platine est le suivant :
m + 2xl + x - q = 10 + 2x2 + (1+3) + 0 = 18
(La structure électronique de la bande de valence du platine est 6s2, 5d8. Le degré d’oxydation du platine est +4.)
Dans le sel noir du Roussin K[Fe4S3(NO)7] on est en présence d’une configuration géométrique originale. Les 4 atomes de fer et les trois ligands X2 :S occupent les sommets d’un cube dont le huitième sommet est vacant. Chaque ligand :S est relié à deux atomes de fer. Une autre particularité de ce complexe est que trois des atomes de fer sont dans la configuration Fe(NO)2S2 et le quatrième (celui qui est diamétralement opposé au sommet vacant) est dans la configuration Fe(NO)S3.

Chélate et denticité
Nous avons vu qu’un même atome au sein d’un ligand pouvait être porteur de plusieurs doublets liants ou de plusieurs électrons non appariés. Un même ligand peut également être porteur de plusieurs atomes susceptibles de se lier à un métal par une liaison de coordination. Le nombre de ces atomes est appelé denticité du ligand. Un ligand peut être bidenté, tridenté, quadridenté... voire même hexadenté. L’exemple le plus connu de ligand bidenté est l’éthylènediammine H2N-CH2-CH2-NH2 (formule condensée C2H8N2). Il peut se coordonner par le biais de ses deux fonctions amine -NH2. Dans le dichloro(éthylènediamine) palladium (II) PdCl2(C2H8N2) l’éthylènediamine se coordonne avec l’atome de palladium par ses deux extrémités amine comme si c’étaient deux ligands L. Un composé complexe au sein duquel l’atome métallique central est relié à un même ligand par deux liaisons de coordination est un chélate.

Le chlorure de dichlorobis(éthylènediammine) cobalt(III) est un sel composé d’un anion chlorure et d’un cation [CoCl2(C2H8N2)2]+. Le cation dichlorobis(éthylènediammine) cobalt(III) a une structure octaédrique : il se coordonne avec deux ligands chlore et deux ligands éthylènediammine par ses deux extrémités amine (ce qui fait 2x2 liaisons de coordination). Il respecte la règle des 18 électrons :
N = 9 + 4x2 + 2 – 1 = 18
L’éthylènediamine tétraacétique C10H16N2O8 (EDTA) est hexadenté : les deux fonctions amine peuvent servir de ligand ainsi que les quatre ions carboxylate (chaque groupe amine est lui-même porteur de deux groupes carboxyliques (goupes -CH2-C(=O)OH). L’EDTA a un pouvoir chelatant fort. On l’utilise comme antidote pour traiter les intoxications par les métaux lourds.
Pour en savoir plus :
post sur la classification périodique des éléments
post sur les liaisons chimiques
post sur les orbitales moléculaires
post sur la valence
post sur la géométrie des molécules
post sur les complexes et les ligands : l’approche ionique
post sur les complexes et les ligands : exemples
post sur l’azote
post sur les acides et les bases
index
#complexe#coordination#chimie#liaison covalente#liaison ionique#chelate#chelation#ligand#denticité#ferrocène#ligand pontant#orbitale moléculaire
0 notes
Text
Electronégativité
L'électronégativité d'un atome est une grandeur qui caractérise sa capacité à attirer les électrons dans une liaison chimique avec un autre élément.
Il existe différents types de liaisons chimiques (voir les posts à ce sujet). Nous allons surtout nous intéresser à la liaison covalente. Dans la liaison covalente, chaque élément met en commun un électron. L’orbitale des deux électrons mis en commun est étendue à la molécule tout entière, ce qui assure la stabilité de la liaison. On pourrait s’attendre à ce que cette orbitale soit répartie de manière symétrique entre les deux atomes de la molécule. C’est en effet le cas lorsque la molécule est formée à partir d’un seul type d’atome (dihydrogène H2, dioxygène O2, diazote N2…). Ce n’est plus nécessairement le cas lorsque ladite molécule est formée à partir d’éléments différents. Prenons le cas de la molécule H2O par exemple. Le noyau d’un atome d’oxygène comporte 8 protons, celui d’un atome d’hydrogène un seul. Les électrons engagés dans la liaison covalente (les électrons de valence) sont plus attirés par le noyau d’oxygène que par le noyau d’hydrogène. Le noyau d’oxygène « tire la couverture à lui ». En clair, cela signifie que la probabilité de trouver les électrons de valence est plus forte près du noyau d’oxygène que des noyaux d’hydrogène. La liaison covalente résultante est partiellement ionique et la molécule d’eau présente un moment dipolaire (on dit qu’elle est polaire).
Pour caractériser cette situation, on dit que l’atome d’oxygène a un potentiel électronégatif plus élevé que l’atome d’hydrogène. La différence d'électronégativité entre les éléments associés dans une liaison covalente permet d’en déterminer la nature :
Lorsque la différence d’électronégativité est faible, on a une liaison covalente apolaire. Les orbitales des électrons de valence sont quasi-symétriques. Les électrons sont attirés de la même façon par les noyaux des atomes.
Une différence d’électronégativité plus forte va entraîner une liaison covalente polaire. La distribution des charges est inégale entre les deux atomes de la molécule. Celle-ci présente un moment dipolaire.
Lorsque la différence est très forte, l'un des atomes attire complètement (ou presque) les électrons engagés dans la liaison qui s’apparente alors à une liaison ionique. Les atomes sont des ions, ils portent une charge et sont retenus par une force principalement électrostatique.
Nota : Il existe un dernier type de liaison covalente que l’on appelle liaison covalente de coordination (ou liaison de coordinence). Dans ce cas le doublet d’électrons qui forme la liaison est apporté par un seul atome.
Echelle de Pauling
Linus Carl Pauling, un chimiste américain, a beaucoup travaillé sur la nature des liaisons chimiques. Il a chercher à les caractériser en fonction des énergies de liaison des atomes. Dans le cas d’une liaison covalente pure, il a constaté que les énergies de liaison des liaisons s’ajoutaient :

Il a donc fait l’hypothèse que tout écart par rapport à cette égalité était le signe d’une dissymétrie dans la liaison (i.e de son caractère partiellement ou complètement ionique) :

De manière empirique, il a vérifié qu’il était possible d’attribuer à chaque atome une grandeur caractérisant son électronégativité, ce qui permet d’écrire :

La différence d’électronégativité s’exprime en kJ/mol. De manière arbitraire, on attribue à l’atome d’hydrogène une électronégativité égale à 2,2 ce qui permet d’établir une échelle d’électronégativité des atomes. Le fluor est l’élément le plus électronégatif de la classification périodique.
Remarque : la formule à laquelle Pauling a abouti est en fait un peu plus complexe, mais le raisonnement reste valable.

La différence d’électronégativité entre les éléments constituant une molécule permet de déterminer la nature de la liaison chimique entre eux. Lorsque la différence d’électronégativité entre ces deux éléments est inférieure à 0,4, la liaison est apolaire. Lorsqu’elle est comprise entre 0,4 et 1,7 elle est polaire. Lorsqu’elle est supérieure à 1,7 on a affaire à une liaison ionique.
Remarque : il existe d’autres formulations de l’électronégativité, dont celle proposée par Robert Mulliken, un autre chimiste américain, qui est basée sur l’affinité électronique et l’énergie d’ionisation des atomes. L’échelle de Mulliken permet d’attribuer une électronégativité aux gaz nobles, ce qui n’est pas le cas avec l’échelle de Pauling.
Dans le tableau de classification périodique, l’électronégativité augmente de la gauche vers la droite sur une même ligne. Elle a tendance à décroître sur une même colonne : le numéro atomique augmentant, les électrons des couches inférieures font écran avec le noyau qui est plus distant des électrons périphériques.
Le tableau qui suit donne les valeurs d’électronégativité pour quelques éléments parmi les plus courants.

On ne sera pas étonné de constater que les halogènes font partie des éléments les plus électronégatifs du tableau. Le fluor, le chlore et le brome partagent avec l’oxygène et l’azote le fait d’avoir une électronégativité supérieure ou égale à 3. De l’autre côté du tableau, l’hydrogène se distingue des autres éléments de la première colonne (les métaux alcalins). Il présente une électronégativité de 2,2 comparable à celle du phosphore, un non-métal de la colonne 15, alors que l’électronégativité des alcalins et de la plupart des alcalino-terreux (dont le calcium) est inférieure à 1. Ceci permet de comprendre l’existence de composés chimiques appelés hydrures dans lesquels c’est l’hydrogène qui est porteur de la charge négative (hydrure de sodium NaH, hydrure de calcium CaH2).
Pour en savoir plus :
post d’introduction à la chimie
post sur les éléments
post sur le nuage électronique
post sur la cohésion de la matière
post sur l’oxydoréduction
post sur le degré d’oxydation
post sur les espèces nucléophiles et électrophiles
post sur les ligands et la complexation
post sur la classification périodique des éléments
glossaire de chimie générale
index
#chimie#électronégatif#ion#covalence#liaison ionique#liaison covalente#oxygène#hydrogène#pauling#mulliken#halogène#alcalin#hydrure
0 notes
Text
Les liaisons chimiques
Il existe différents types de liaisons chimiques. Elles expliquent la plupart des propriétés physiques (cohésion, caractéristiques mécaniques, électriques et thermiques, couleur et transparence) des matériaux, depuis la forme des flocons de neige jusqu’à la cohésion des rochers et des cristaux ou les propriétés électriques des métaux. Toutes les liaisons chimiques ont une origine électrique. Les chimistes identifient six grandes catégories de liaisons chimiques, ou disons plutôt six modèles car la nature brouille souvent les pistes en empruntant des caractéristiques à plusieurs modèles :
la liaison covalente,
la liaison ionique et la liaison iono-covalente,
la liaison métallique,
la liaison hydrogène, ou pont hydrogène, et la liaison halogène qui est de nature assez semblable,
les forces de Van der Waals (forces de Keesom, forces de Debye, forces de London).
Liaison covalente
Nous n’insisterons pas sur ce type de liaison auquel sont consacrés deux posts détaillés (approche simplifiée et théorie des orbitales moléculaires)..En résumé, les électrons d'un atome évoluent dans une région de l'espace appelée orbitale. La forme de cette orbitale est déterminée par une équation de la mécanique quantique, l'équation de Schrödinger. Les différentes orbitales sont les solutions de cette équation appliquée à un potentiel colombien représentatif du noyau. Dans le cas d'une molécule, la présence de plusieurs noyaux modifie ce potentiel coulombien. Il apparaît des orbitales moléculaires qui s'étendent sur plusieurs atomes et dans lesquelles ceux-ci mettent en commun des électrons de leur couche périphérique (la bande de valence). L'énergie de ces orbitales moléculaires étant plus faible que celles des atomes d'origine, la liaison est stable. Le différentiel d'énergie est appelé énergie de liaison. La molécule d’eau est un exemple de liaison covalente. Elle intervient également dans la constitution de certains cristaux.
Liaison ionique
La liaison ionique (aussi appelée liaison électrovalente) est une liaison basée sur les forces d’interaction électrostatique. Elle se forme entre deux éléments ayant un potentiel électronégatif très différent. L’élément le plus électronégatif capte un ou plusieurs électrons de l’autre élément (le plus souvent un métal) et il se forme deux ions. L'électronégativité est la capacité d'un élément chimique à attirer les électrons lors de la formation d'une liaison chimique avec un autre élément. La différence d'électronégativité entre ces deux éléments s’exprime en électron-volts (eV). Elle mesure le delta d’énergie lié au captage d’un électron. On estime qu’il faut un différentiel d’électronégativité de 1,7 eV pour former une liaison ionique.
Dans le cas du chlorure de sodium NaCl par exemple (le sel de table), le sodium possède un seul électron de valence alors que le chlore en a sept. L’atome de sodium est moins électronégatif que l’atome de chlore et il lui cède son électron de valence pour former une liaison ionique :

Sur le plan énergétique, le fait de retirer un électron au sodium est endothermique (requiert de l’énergie). L’addition d’un électron à l’atome de chlore est quant à elle exothermique et le bilan est favorable.
Ce que nous avons décrit à l’échelle de deux atomes peut se produire à une échelle moléculaire. Par exemple, le sulfate d’ammonium (NH4)2SO4 (un engrais) est composé de deux cations NH4+ (ammonium) et d’un anion SO42- (sulfate).
Dans un composé à liaison ionique, la symétrie sphérique de la distribution électronique de chaque ion est conservée (les électrons restent localisés sur les ions et ne sont pas partagés). La liaison ionique est donc non directionnelle. C’est également une liaison forte : environ 5eV par paire d’atomes liés.
La liaison ionique permet le formation de cristaux. C’est d’ailleurs sous cette forme (ou plus exactement sous la forme de microcristaux) que se présentent la plupart des sels. Prenons par exemple le cas d’un bloc de calcite. La calcite est un cristal de carbonate de calcium Ca2+CO32-. Mais il n’y a pas, à proprement parler, de molécule CaCO3 dans ce réseau cristallin. Dans un cristal de calcite, les cations Ca2+ et les anions polyatomiques CO32- sont positionnés de manière régulière et ordonnée de façon à ce que les forces électrostatiques qui s’appliquent sur chacun d’eux s’équilibrent. C’est cet équilibre qui conduit à la formation de microcristaux... ou même de cristaux. Le rubis et le saphir, des pierres précieuses très utilisées en joaillerie, sont des cristaux d’alumine Al2O3 auxquels la présence d’oxyde donne leur couleur. Comme dans le cas de la calcite, la nature des liaisons au sein du cristal est ionique. Il y a deux cations Al3+ pour trois anions O2-.
La liaison iono-covalente est une liaison covalente polarisée. Une liaison covalente entre deux atomes de nature différente (et plus particulièrement d’électronégativité différente) ne peut pas être symétrique. Dans ce cas la distribution électronique de l’orbitale moléculaire qui supporte la liaison est décalée vers l’atome de plus forte électronégativité, ce qui donne un caractère partiellement ionique à la liaison. C’est le cas par exemple pour la molécule d’eau. Les électrons covalents ont plus de chance de se trouver près de l’atome d’oxygène qu’à proximité des atomes d’hydrogène et il en résulte la création d’un dipôle électrique. Les molécules comme l’eau qui présentent un dipôle électrique sont dites polaires.
Au demeurant, seuls les éléments très nucléophiles ou très électrophiles (alcalins, alcalino-terreux, chalcogènes et halogènes) sont de nature à capter – ou donner – un ou deux électrons. En dehors de ces éléments, le transfert de la charge n’est souvent que partiel. Même dans le cas de liaisons réputées ioniques il peut y avoir un caractère partiellement covalent.
Liaison métallique
La liaison métallique tire sa spécificité d’une propriété physique des éléments chimiques que l’on appelle les métaux. Dans le cas des métaux, la bande de valence (les niveaux d’énergie des électrons de valence) et la bande de conduction (les niveaux d’énergie que peuvent occuper les électrons libres) sont connexes. Elles ne forment en fait qu’une seule bande et ceci permet à un fluide d’électrons libres délocalisés de se former dans le métal (ou l’alliage). Les cations métalliques forment un réseau tridimensionnel dont la cohésion est assurée par ce fluide. La liaison métallique est non directionnelle. Elle est relativement forte : environ 1 eV par paire liée.
L’existence de ce fluide d’électrons fait des métaux et des alliages de bons conducteurs thermiques et électriques. La nature de cette liaison, qui est moins rigide que les précédentes, donne par ailleurs aux métaux et aux alliages leur malléabilité et leur plasticité. Cette liaison se maintient en effet lors de déformations alors que les liaisons covalentes ou ioniques se brisent.
Comme dans le cas de la liaison covalente, il n’y a pas de liaison métallique pure : les atomes métalliques forment aussi entre eux des liaisons covalentes (dites de coordination), ce qui explique la structure cristalline que l’on peut observer à l’échelle granulaire dans les métaux à l’état solide.
La liaison métallique ne s’applique pas qu’aux métaux. Il existe un état de l’hydrogène appelé hydrogène métallique dans lequel les atomes d’hydrogène partagent leur unique électron. On suppose que les couches internes des étoiles gazeuses sont occupées par de l’hydrogène métallique.
Pont hydrogène
Comme nous l’avons vu au paragraphe concernant la liaison iono-covalente, les liaisons covalentes peuvent donner lieu à la formation d’un dipôle en raison de la différence de potentiel électronégatif entre les deux éléments engagés dans la liaison. Ceci permet la création de liaisons entre ces dipôles. C’est le cas par exemple de l’eau à l’état solide et c’est ce qui lui donne le caractère cristallin de la glace (ou des flocons de neige). On appelle ce type de liaison une liaison hydrogène (ou pont hydrogène). Cette liaison ne peut se produire qu’entre un atome d’hydrogène et un élément fortement électronégatif comme l’oxygène, l’azote et le fluor.
Les liaisons hydrogène jouent un rôle important dans la cohésion de composés macromoléculaires comme les polymères, qu’ils soient de synthèse comme les polyamides ou les polyuréthanes, ou d’origine naturelle comme les protéines ou la cellulose. Dans le cas du papier, ce sont également des ponts hydrogène qui maintiennent ensemble les fibres de cellulose qui le constituent.
La liaison hydrogène est classée parmi les liaisons faibles : 0,1 eV par paire liée. Elle est directionnelle. La liaison halogène est de même nature que la liaison hydrogène. Dans ce cas, c’est un élément halogène (astate, iode, brome ou chlore) qui joue le « rôle du cation ».
Forces de Van der Waals
Les forces de Van der Waals regroupent divers types de forces électrostatiques de plus faible intensité. Elles ont été découvertes par Johannes Diderik Van der Waals et lui ont valu le prix Nobel en 1910. Il en existe de différents types : force de Keesom, force de Debye, force de London. Elles sont dues à l’interaction entre des dipôles électriques, permanents ou induits. Elles sont non directionnelles et de faible intensité (quelques meV par paire liée).
On les rencontre dans des structures cristallines en feuillets ou en lamelle. C’est le cas pour le graphite. La structure du graphite est constituée de la superposition de feuillets de structure hexagonale décalés les uns par rapport aux autres. Les liaisons entre atomes de carbone d’un même feuillet sont covalentes. La cohésion entre feuillets est assurée par des forces de Van der Waals. Le nombre important d’atomes dans chaque feuillet compense la faiblesse de ces forces. (Il n’en reste pas moins que le graphite est beaucoup plus friable que le diamant !)
Les forces de Van der Waals assurent également la cohésion des matériaux polymères amorphes ou semi-cristallins.
Si ces forces sont d’intensité nettement moindre que les précédentes, elles ne sont cependant pas négligeables… Ce sont les forces de Van der Waals qui permettent au gecko de grimper et de se maintenir sur des parois de verre verticales !
Panachage
D’une manière générale, la formation d’assemblages moléculaires (et en particulier de cristaux) ne relève pas d’un seul type de liaison. Elle résulte de l’existence d’un optimum énergétique lié à une configuration géométrique, à caractère périodique, localement ou amorphe, dans laquelle peuvent intervenir des liaisons covalentes, électrovalentes, iono-covalentes, des ponts hydrogène et des forces de Van der Waals. Le gypse CaSO4.2H2O en est un exemple emblématique. Il est constitué de feuillets de CaSO4 maintenus entre eux par des forces de Van der Waals qui sont véhiculées par les molécules H2O situées dans les espaces interfoliaires. Au sein de ces feuillets, les anions SO42- ont une structure tétraédrique centrée de nature plutôt covalente (les liaisons sont de nature directionnelle). Chaque cation Ca2+ est relié à plusieurs anions, les liaisons étant cette fois ioniques, et à deux molécules d’eau par des forces de Van der Waals !
Pour en savoir plus :
post sur la cohésion de la matière
post sur la structure du nuage électronique
post sur la valence
post sur la liaison covalente (approche simplifiée)
post sur les orbitales moléculaires
post sur les cristaux
post sur les cristaux (suite)
post sur les composés complexes
post sur la cohésion de la matière
post sur les roches sédimentaires
post sur la classification périodique des éléments
glossaire de chimie générale
index
#chimie#covalence#liaison covalente#liaison ionique#liaison métallique#van der waals#pont hydrogène#iono-covalente#covalente#ionique#électrovalente#liaison sigma
0 notes
Text
La cohésion de la matière solide
Faire de la physique, ce n’est pas nécessairement se poser des questions compliquées sur des phénomènes étranges. C’est aussi, très souvent, se poser des questions toutes bêtes, comme par exemple : Pourquoi le ciel est bleu ? Pourquoi la nuit est noire ?
Alors demandons-nous aujourd’hui pourquoi la matière est solide. Qu’est-ce qui fait que les rochers sont de gros blocs ? Pourquoi je me fais mal lorsque je me cogne contre un mur ? Et, accessoirement, pourquoi le verre est transparent et pas le métal… La question, au regard de nos connaissances actuelles au sujet des atomes, n’est pas triviale. Nous savons depuis le début du XXème siècle et les expériences de Rutherford que la matière est principalement composée de vide. La distance moyenne entre atomes est de l’ordre de l’Angström (10-10 m) alors que la dimension d’un noyau atomique n’excède pas 10-15 m ! Comment des atomes isolés, apparemment isolés les uns des autres et de nature parfois très différente peuvent-ils former un bloc solide ? Pourquoi ne passe-t-on pas au travers d’un tel bloc ?
Pour résoudre cet apparent paradoxe, il nous faut nous déshabituer à considérer les atomes comme de petites billes neutres électriquement. Un atome est constitué d’un noyau, chargé positivement et localisé en son centre, et d’un nuage électronique qui l’entoure et dont la meilleure représentation est une somme de densités de probabilité de présence des électrons (ce qui revient à supposer une densité de charge électrique négative). La structure du nuage électronique résulte à la fois d’un optimum énergétique pour le champ des électrons dans le potentiel coulombien du noyau et d’une condition de stationnarité du champ électromagnétique des électrons. Si on remplace le potentiel coulombien à symétrie sphérique d’un noyau isolé par une multitude de puits de potentiel correspondant à une multitude de noyaux, il est clair que les conditions qui conduisent à l’apparition d’un optimum énergétique pour un ensemble de champs stationnaires peuvent conduire à une solution tout à fait différente. C’est en particulier le cas si la répartition des noyaux est périodique dans une, deux ou trois dimensions (apparition d’interférences). C’est ce qui conduit à la formation de cristaux... et plus généralement à la cohésion de la plupart des solides. Lorsque le nouvel optimum énergétique est plus favorable, c’est-à-dire lorsqu’il permet aux électrons d’abaisser leur niveau d’énergie, les matériaux en se refroidissant vont avoir tendance à s’agréger pour former une matière solide dont les propriétés mécaniques vont dépendre de la nature des liaisons qui se sont formées.
Liaisons chimiques
A cet optimum énergétique est associé la notion de liaison chimique. Une liaison chimique est une interaction qui maintient deux ou plusieurs atomes ou molécules à courte distance les uns des autres. L’énergie totale des atomes, des molécules ou des ions qui entretiennent une liaison chimique est plus faible que celle de ces mêmes atomes, molécules ou ions dispersés. Il faut donc fournir une certaine énergie pour les séparer. On appelle cette énergie l’énergie de liaison. Cette énergie est exprimée en eV (électron-Volt). Il existe plusieurs types de liaisons chimiques. Les chimistes en distinguent six, classés généralement par ordre décroissant des énergies de liaison :
la liaison covalente, qui peut conduire à des molécules polaires ou apolaires en fonction des potentiels électronégatifs respectifs des atomes concernés,
la liaison ionique,
la liaison iono-covalente,
la liaison métallique,
la liaison hydrogène, ou pont hydrogène, et la liaison halogène qui est de nature assez semblable,
les forces de Van der Waals (forces de Keesom, forces de Debye, forces de London).
La liaison covalente est une liaison directionnelle qui résulte de la mise en commun d’électrons. C’est une liaison forte : supérieure à 5 eV par paire liée. La liaison covalente de coordination peut être considérée comme une forme de liaison covalente. Elle trouve son explication dans la formation d’orbitales moléculaires tout comme la liaison covalente simple.
La liaison ionique est une liaison électrostatique entre ions de charges opposées. Elle n’est pas directionnelle. C’est également une liaison forte (~5 eV par paire). La liaison iono-covalente consiste en un mix des deux, lorsque les électrons qui assurent la liaison sont attirés par l’un des pôles sans que l’on puisse toutefois parler d’ionisation. La liaison métallique est non directionnelle et d’intensité moyenne (~1 eV par paire). Elle est assurée par un flux d’électrons libres qui circulent dans un réseau cristallin.
La liaison hydrogène est une liaison faible (0,1 eV) assurée par un atome d’hydrogène entre deux atomes électronégatifs. Les forces de Van der Waals sont des forces qui se créent entre atomes ou molécules en raison de l’interaction entre leurs moments dipolaires instantanés (fluctuations quantiques de la densité électronique autour des noyaux) et les moments dipolaires qu’ils induisent. Les forces de Van der Waals sont les plus faibles, le gain énergétique par paire n’est que de quelques meV par paire liée.
Structure de la matière
La cohésion de tous les solides repose sur ces liaisons. Elles peuvent conduire à des matériaux très structurés comme les cristaux ou à des matériaux dans lesquels les molécules ou les atomes sont distribués de manière aléatoire (matériaux amorphes). Les molécules composant un solide peuvent également constituer un réseau formé par la répétition d’un ou plusieurs motifs élémentaires, on parle alors de polymère. La molécule constituant le maillon élémentaire d’un polymère est appelée monomère. Les polymères peuvent former des nappes ou des fibres. Les métaux forment une classe à part du fait de la liaison métallique.
Ces différentes formes d’association peuvent se combiner. Un alliage par exemple est généralement constitué d’un agrégat de petits cristaux métalliques.
Propriétés de la matière
C’est la combinaison de ces liaisons chimiques qui donne à la matière la plupart de ses propriétés mécaniques. Tout d’abord sa dureté. Nous nous demandions au début de ce post pourquoi ne peut-on pas passer au travers d’un mur. Prenons deux blocs de matière solide, qu’ils soient de nature différente ou identique, amorphe (composée d’atomes reliés entre eux de façon désordonnée) ou cristalline. Comme nous l’avons dit plus haut, les atomes qui composent ces blocs sont reliés entre eux par des liaisons chimiques. Quel que soit le type de liaison, elle assure à l’ensemble un état d’énergie minimale. Les faire s’interpénétrer reviendrait à modifier entièrement les champs électriques qui assurent la cohésion de l’assemblage, et donc à casser les liaisons existantes. La matière va s’opposer violemment à cette tentative d’interpénétration. Si les matériaux sont ductiles on assistera à une déformation des matériaux au contact. Si la contrainte est trop forte, l’un des blocs va se rompre (résistance). Ou alors il est possible que l’un des deux corps perfore l’autre. Toutes ces propriétés dépendent directement de la nature et de l’intensité des liaisons entre les atomes.
Les caractéristiques électriques de la matière dépendent aussi en grande partie de ces liaisons. Dans un cristal covalent, les atomes de valence sont piégés par les liaisons covalentes. Le matériau sera donc isolant (voir semi-conducteur si le cristal est dopé par des impuretés, voir le post sur les semi-conducteurs). Dans le cas d’un métal, un fluide d’électrons libres assure la liaison entre les atomes et garantit une forte conductivité. Idem pour les caractéristiques thermiques, tant du point de vue dilatation que conductivité et point de fusion (liée à l’énergie de liaison).
Couleur et transparence
La transparence de la matière dépend de sa capacité à interagir avec la lumière dans le domaine visible. Dans le cas d’un diamant par exemple, les électrons de valence sont tous piégés par les liaisons covalentes. Ils n’interagissent pas avec la lumière visible qui passe facilement au travers du cristal. Idem pour le corindon monocristallin sans impureté. Cette fois ce sont les liaisons ioniques qui emprisonnent les électrons de valence dans leur réseau. C’est la présence d’oxyde de chrome qui donne au rubis (une pierre précieuse à base de corindon) ses reflets rouges. N’étant pas prisonnier des liaisons ioniques, l’oxyde de chrome interagit avec la lumière et réémet de la lumière rouge. Idem pour l’émeraude, une pierre précieuse à base de silicate aux reflets verts dus à la présence de trace de chrome et de vanadium.
Dans le cas des métaux, c’est l’inverse. Le fluide d’électrons délocalisés bloque toute pénétration par les ondes électromagnétiques sauf dans les rayons X (longueur d’onde inférieure à la distance interatomique). Les métaux sont parfaitement opaques : la lumière incidente est réfléchie par la surface (réflexion spéculaire). Le métal poli finement (taille des défauts inférieure à la longueur d’onde) présente même l’aspect d’un miroir. La couleur des métaux est en général blanche ou grise (argentée) sauf pour l’or et le cuivre.
Dans le cas des autres matériaux, l’onde électromagnétique pénètre plus ou moins profondément et interagit avec les atomes et les électrons. C’est la lumière réémise par ceux-ci qui donne leur couleur auxdits matériaux.
Les différentes formes de la matière solide
Cristal
Un cristal est composé d’un assemblage d’atomes ou d’ions qui présente une triple périodicité (trois mailles élémentaires orientées dans trois directions différentes). La structure cristalline est très répandue dans la nature. On associe souvent la notion de cristal à des matériaux nobles comme le diamant. Le diamant constitue d’ailleurs un très bon exemple pour comprendre la nature d’un cristal. Il est composé exclusivement d’atomes de carbone. Le carbone est tétravalent : il peut entretenir 4 liaisons covalentes. Dans le diamant, les atomes de carbone se placent de façon à occuper les sommets et le centre des faces d’un cube. Quatre autres atomes complètent cet arrangement de façon que chaque atome de carbone entretienne quatre liaisons covalentes avec ses voisins le plus proches.

Les cristaux covalents sont très rigides et très robustes. De nombreux outils de perçage ou de coupage en mécanique ont une pointe en diamant.
Les cristaux ne sont pas tous basés sur des liaisons covalentes. La liaison ionique permet, tout comme la liaison covalente, la constitution de cristaux. Ce type de cristaux est cependant de nature tout à fait différente. Dans un cristal covalent, les atomes sont solidement et rigidement reliés les uns aux autres par des orbitales communes qui assurent un caractère directionnel à la liaison.
Ce n’est pas le cas dans un cristal composé d’ions salins. La liaison ionique n’est pas directionnelle. Prenons par exemple le cas d’un bloc de calcite. La calcite est un cristal de carbonate de calcium Ca2+CO32-. Mais il n’est pas composé de molécules CaCO3. Il n’y a d’ailleurs pas, à proprement parler, de molécule CaCO3 dans le réseau cristallin. Dans un cristal de calcite, les cations Ca2+ et les anions polyatomiques CO32- sont positionnés de manière régulière et ordonnée de façon à ce que les forces électrostatiques qui s’appliquent sur chacun d’eux s’équilibrent.
L’énergie de liaison ionique est en général un peu plus faible que l’énergie de liaison covalente. Il ne faut cependant pas s’y tromper : si le sel de table ou le calcaire s’écrasent facilement, le corindon (cristal d’alumine anhydre Al2O3) est quant à lui très dur. Sous forme rocheuse, le corindon porte le nom d’émeri, une pierre connue pour ses propriétés abrasives. (Le rubis et le saphir sont des pierres précieuses dont la couleur est due à la présence d’oxydes dans un cristal de corindon.)
La glace est un autre exemple de solide cristallin. Mais cette fois il n’y a ni liaison covalente ni liaison ionique. La nature cristalline de l’eau à l’état solide est due à la liaison hydrogène (ou pont hydrogène) rendue possible par le caractère polaire de la molécule d’eau. Les atomes d’oxygène polarisés négativement attirent les atomes d’hydrogène polarisés positivement et il en résulte un arrangement sous forme de cristal qui permet à la glace de minimiser son état énergétique.

D’une manière générale, la formation de cristaux n’est pas spécifique à un type de liaison donné. Elle résulte de l’existence d’un optimum énergétique lié à une configuration géométrique à caractère périodique dans laquelle peuvent intervenir des liaisons covalentes, électrovalentes, ionocovalentes, des forces de Van der Waals ou des ponts hydrogène. Le gypse CaSO4.2H2O par exemple est constitué de feuillets de CaSO4 maintenus entre eux par des forces de Van der Waals véhiculées par les molécules H2O. Au sein de ces feuillets, les anions SO42- ont une structure tétraédrique centrée de nature plutôt covalente (les liaisons sont de nature directionnelle). Chaque cation Ca2+ est relié à plusieurs anions, les liaisons étant cette fois plutôt ioniques, et à deux molécules d’eau par des forces de Van der Waals !
Il existe plusieurs structures de réseau cristallin en fonction de la position des atomes ou molécules qui le constituent et du nombre de liaisons entre eux. Les plus connues sont les structures cubiques (cubique simple, cubique centrée, cubique à faces centrées) et hexagonales (orthorhombique, clinorhombique, rhomboédrique…). La cristallographie est la science qui décrit les propriétés physico-chimiques des cristaux en faisant le lien avec leur géométrie à l’échelle atomique.
Remarque : la forme cristalline la plus répandue est en fait le polycristal, formé par l’agrégation de petits cristaux (cristallites) d’orientation arbitraire. Leurs propriétés mécaniques dépendent en grande partie de leur microstructure, c’est-à-dire de la taille des cristallites et de la structure des joints entre les grains (intermétallique).
Métaux et alliages
La structure des éléments classés dans la catégorie des métaux tire sa spécificité de la liaison métallique. La liaison métallique repose sur une propriété physique des métaux. La bande de valence et la bande de conduction des métaux (les niveaux d’énergie des électrons de valence et les niveaux d’énergie que peuvent occuper les électrons libres) sont connexes. Elles ne forment en fait qu’une seule bande et ceci permet à un fluide d’électrons libres délocalisés de se former dans le métal (ou l’alliage). Les cations métalliques forment ainsi un réseau tridimensionnel dont la cohésion est assurée par ce fluide.
Ce type de liaison n’exclut cependant pas l’existence de liaisons covalentes de coordination (mise en commun de doublets d’électrons) entre atomes voisins. Ceci conduit à une structuration en cristaux à une échelle microscopique. A l’état solide, métaux et alliages sont donc composés de grains cristallins de taille plus ou moins grande agrégés entre eux. On parle de solide polycristallin. Dans la plupart des cas, la structure cristalline de ces grains est soit cubique centrée, soit cubique à faces centrées soit hexagonale compacte. Lorsque le métal ou l’alliage se solidifie, de nombreux cristaux se forment dans le liquide. Le moment où les cristaux commencent à croître est appelé nucléation. Très souvent, la cristallisation est initiée par la présence d’impuretés. Lorsque la température diminue, les atomes métalliques se déposent sur ces impuretés, ce qui favorise la croissance de cristaux. A mesure que ces cristaux grossissent ils en viennent à se toucher. Il se forme alors des joints entre les grains cristallins (joints de grain). La zone du joint de grain est formée d’atomes qui n’ont pas de structure cristalline (zone désordonnée). Ils participent néanmoins à la liaison métallique, ce qui assure la cohésion de l’ensemble.
L’existence du fluide d’électrons responsable de la liaison métallique fait des métaux et des alliages de bons conducteurs thermiques et électriques. La nature de cette liaison, qui est moins rigide que les précédentes, donne par ailleurs aux métaux et aux alliages leur malléabilité et leur plasticité. Cette liaison se maintient en effet lors de déformations alors que les liaisons covalentes ou ioniques se brisent.
Matériaux amorphes
Les matériaux amorphes sont des solides au sein desquels, les molécules ou atomes ne sont pas ordonnés suivant une structure définie mais sont aléatoirement distribués comme dans un liquide. Ils sont obtenus par refroidissement rapide d’un liquide (la trempe). Au cours de cette trempe, les atomes ou molécules n’ont pas le temps de cristalliser et restent bloqués dans une configuration correspondant localement à un optimum énergétique. Ils occupent donc une position fixe. Il y a eu transition vitreuse.
Remarque : le verre est un exemple de matériau amorphe. Il est composé en grande partie de silice. Lorsqu’on refroidit la silice lentement, elle cristallise sous la forme de cristaux de quartz. Lorsqu’on la trempe, elle donne du verre.

Les matériaux amorphes sont dans un état métastable : cet état ne correspond pas à l’optimum énergétique. Mais l’agitation thermique est largement insuffisante pour permettre aux molécules de se déplacer et de s’orienter de façon à former un cristal. L'augmentation progressive de la température conduit les molécules ou les atomes à devenir de plus en plus mobiles. Le matériau passe alors progressivement d'un état amorphe solide à un état liquide de haute viscosité sans qu’on puisse déterminer une température de fusion précise.
Il existe plusieurs types de matériaux amorphes et on peut les classer en trois grandes catégories : les verres, les polymères organiques et les élastomères. On donne le nom de verre de manière générique aux matériaux transparents. De façon plus restrictive on l’applique aux matériaux amorphes composés de molécules minérales. Dans le verre industriel par exemple, la composante dominante est la silice SiO2. A l’état naturel la silice est généralement sous forme cristalline. La trempe de la silice en fusion et l'ajout d'additifs qui limitent la formation de cristaux permettent d'obtenir le verre tel qu’on le connaît. Lorsque le matériau obtenu est exempt de microcristaux, on lui donne le nom de gel de silice. En règle générale, le verre comporte des microcristaux noyés dans une pâte amorphe. Les liaisons chimiques au sein des verres sont le plus souvent de type ionique ou iono-covalente.
Un polymère organique amorphe est un matériau composé de macromolécules (polymères) repliées et enchevêtrées. Dans le cycle de fabrication de ces matériaux, lors du passage de la phase liquide à la phase solide, les macromolécules peinent à s’orienter pour former des cristaux. La solidification se produit lorsqu’elles ne peuvent plus bouger. C’est le cas en particulier lorsque la variation de température est brutale (processus similaire à la trempe des métaux ou du verre). Comme dans le cas des verres, ce type de matériau est partiellement cristallin en ce sens qu’il comporte des amorces de cristaux. Les élastomères sont des matériaux à base de polymères organiques dans lesquels certaines macromolécules sont pontées entre elles (par exemple par des chaînes soufrées) pour former un réseau tridimensionnel (on parle de réticulation).
Les liaisons chimiques entre polymères organiques sont généralement des liaisons chimiques faibles (pont hydrogène ou forces de Van der Waals). Les matériaux amorphes sont souvent transparents ou translucides. La couleur d'un matériau est la plupart du temps le produit de l’interaction entre les ondes électromagnétiques incidentes et les liaisons interatomiques au sein d’un réseau cristallin. Le caractère majoritairement amorphe du matériau fait qu’une telle interaction ne se produit pas (à moins d’ajouter un colorant). Dans le cas de polymères organiques amorphes, on parle de verres thermoplastiques. C’est le cas par exemple du PMMA (polyméthacrylate de méthyle) ou de certains polycarbonates.
Les roches
Dans l’inconscient collectif, la roche est l’élément associé le plus souvent à la notion de solidité. Ne dit-on pas solide comme un roc ? Les roches sont constituées de grains cristallins de taille plus ou moins grande prisonniers d’une pâte amorphe appelée ciment. Classiquement, on distingue trois types de roches :
les roches magmatiques,
les roches métamorphiques,
les roches sédimentaires.
Les roches magmatiques (on dit aussi ignées) sont, comme leur nom l’indique, issues du magma qui remonte en surface lors d’éruptions volcaniques. La taille des grains cristallins dépend la vitesse de refroidissement du magma. Inférieurs à 2 mm on les appelle des microlithes ou des sphérulites. Les roches contenant ce types de microcristaux sont dites microlithiques. Au-delà de 2 mm on dit qu’elles sont grenues. Le granite, le basalte, la péridotite ou l’andésite sont des roches magmatiques. C’est leur origine et leur texture qui les distinguent beaucoup plus que leur composition minéralogique (olivine, amphibole, pyroxène…).
Les roches sédimentaires forment l’essentiel des roches présentes dans la croûte terrestre. Elles sont produites à l’issue d’un long cycle combinant transformation et transport de matière. La première phase est due à l’altération superficielle de roches par érosion, ruissellement, gel… Cette altération produit des particules et des poussières qui sont transportées par le vent ou les cours d’eau jusqu’à un lieu où elles se déposent et s’accumulent (sédimentation). La dernière phase est la diagénèse. Le terme de diagénèse recouvre divers processus mais tous conduisent à la cimentation des microcristaux (ou des grains) entre eux. La cimentation peut être précoce. L’eau saturée de minéraux circule entre les grains. Les minéraux précipitent sur les grains et finissent par les souder lorsque l’eau se retire. Elle peut être plus tardive et liée à la compaction des sédiments : les minéraux présents entre les grains soudent ceux-ci entre eux du fait de la pression. Les grès sont des exemples types de roches sédimentaires. On notera que certaines roches sédimentaires sont obtenues directement par précipitation. C’est la cas de la dolomie et du gypse.
Les roches métamorphiques sont issues de la transformation de roches ignées ou sédimentaires sous l'effet de température et (ou) de pressions élevées. Le granite par exemple est transformé en schiste ou en gneiss, le grès en quartzite et le calcaire en marbre. L’ardoise quant à elle provient de la transformation métamorphique de boue appelé mudstone.
Les liaisons chimiques qui assurent la cohésion des roches sont presque exclusivement de nature covalente, ionique ou iono-covalente.
Céramiques
Les céramiques forment une autre classe de matériaux. Elles sont composées d’éléments métalliques mêlés à des substances non métalliques (oxydes, nitrures, silicates). Elles peuvent exister sous une forme cristalline ou amorphe, ou comme une combinaison des deux. La cohésion des céramiques est assurée par des liaisons ioniques ou iono-covalentes. Ce sont des matériaux durs et résistants à l’usure mais cassants. Dans les céramiques, les anions sont très souvent différents en taille des cations. La silice (SiO2) et surtout l’alumine (Al2O3) sont à la base de beaucoup de céramiques. Le nitrure de silicium (Si3N4) et le nitrure d’aluminium (AlN) sont à la base de céramiques dites non oxydées.
Polymères, résines et mousses
Nous ne nous attarderons pas sur les polymères : on se reportera à ce sujet aux posts consacrés à la chimie organique.
Les polymères sont à la base des fibres végétales (et donc du bois). Les résines sont composées de polymères renforcés par des fibres ou des billes (on parle dans ce cas de matière plastique). Une mousse est un matériau continu comportant des microbulles de gaz dans de la matière condensée (le plus souvent une résine). Les mousses sont obtenues par solidification (refroidissement ou polymérisation) de mousses liquides. Une mousse comporte au minimum 70% de gaz en volume.
Pour en savoir plus :
post sur les liaisons chimiques
post sur les électrons dans les liaisons chimiques
post sur les roches sédimentaires
post sur la classification périodique des éléments
post sur les sels
post sur les complexes et la liaison de coordination
post sur les propriétés des hydrocarbures
post sur les semiconducteurs
glossaire de chimie générale
index
#physique#chimie#matière#liaisons chimiques#liaison covalente#liaison ionique#van der waals#pont hydrogène#polymère#mousse#cristal#diamant#céramique#cohésion#roche#métamorphique#sédimentaire#granite#basalte#grès#métal#électron#covalence
0 notes
Text
Complexation, composé de coordination et ligand
La notion de composé complexe est indispensable pour comprendre les propriétés de très nombreux corps chimiques. Elle n’est pas intuitive : elle est moins simple à comprendre que celle de liaison covalente ou de liaison ionique. Nous allons dans un premier temps définir ce qu’est un complexe et une liaison de coordination. Nous complèterons cette définition par quelques règles de base. Dans des posts ultérieurs, nous ferons état d’une autre approche de la liaison de coordination et nous donnerons un certain nombre d’exemples illustrant la richesse de cette branche de la chimie.
Un complexe est un composé chimique polyatomique constitué autour d'un corps central (le plus souvent un cation métallique) entouré de plusieurs ligands. On appelle ligand une molécule ou un ion qui possède dans sa bande de valence des doublets non liants dont l'orbitale peut être étendue de façon à englober le corps central, formant ainsi avec lui une liaison chimique. On appelle liaison de coordination ce type de liaison.
Remarque : nous verrons dans le post suivant que certains ligands organiques peuvent former des complexes à partir d’un doublet liant (notion d’hapticité).
Orbitales moléculaires
La notion de liaison de coordination peut s'expliquer dans le cadre de la théorie des orbitales moléculaires. C'est la mécanique quantique et le concept de fonction d'onde qui permettent le mieux de comprendre la structure du nuage électronique autour d'un atome. Les électrons d'un atome ne peuvent occuper que certains niveaux d'énergie correspondant à des formes d'ondes déterminées. Ces formes d'onde sont les seules solutions stationnaires de l'équation de Schrödinger autour de cet atome. (Dans le cas de l'atome d'hydrogène, ce sont des harmoniques sphériques.) On donne à ces formes d'onde le nom d'orbitales atomiques (OA).
Au sein d'une molécule, on montre qu'il existe aussi des solutions stationnaires de l'équation de Schrödinger pour les électrons de valence, solutions qui peuvent s'exprimer sous la forme d'une combinaison linéaire des formes d'onde des OA des atomes qui constituent cette molécule. Une telle orbitale est appelée orbitale moléculaire (OM). Une OM peut donner lieu à une liaison chimique si elle permet au doublet d'électrons qui l'occupent d’avoir un niveau d’énergie inférieur.
Dans le cas d'une liaison covalente standard, chaque atome contribue à part égale à l'occupation de l'OM. Dans la molécule dihydrogène H2 par exemple, chaque atome d'hydrogène place un électron dans cette OM. La cohérence de la molécule O2 quant à elle est assurée par deux OM et chaque atome d'oxygène place un électron dans chacune d'entre elles, ce qui fait deux doublets au total.
Dans une liaison entre un cation métallique et un ligand, c'est le ligand qui fournit le doublet qui occupe l'orbitale moléculaire. Le cation se contente de fournir le site de coordination, le puits de potentiel qui conditionne la géométrie de l'orbitale moléculaire. C’est pourquoi on utilise aussi le terme de liaison dative (bien qu’il soit considéré aujourd’hui comme obsolète) : le ligand « donne » une paire d’électrons pour constituer la liaison, le cation est accepteur de la liaison.
Liaison cation métallique-ligand
De nombreux composés complexes se forment autour de cations de métaux de transition. Les métaux de transition sont des éléments du bloc "d" de la classification périodique des éléments. Ils occupent la rangée 4, 5 ou 6 du tableau périodique des éléments et leur bande de valence a une configuration électronique du type (n+1)s2, ndx avec x inférieur à 10 (sous-couche d incomplète). Les éléments de la 7ème rangée ainsi que le lanthane La ne font pas partie de la famille des métaux de transition bien qu'appartenant au bloc d. Le zinc Zn, le cadmium Cd et le mercure Hg ont quant à eux leur sous-couche d remplie et des propriétés chimiques très différentes.
Remarque : certains métaux dits de post-transition (ou métaux pauvres) comme l’aluminium, le gallium, l’étain, le plomb ou l’indium, sont également susceptibles de former des composés complexes.
Les orbitales moléculaires des complexes cations-ligands résultent d'une combinaison linéaire entre une orbitale du ligand et l'une des orbitales (n+1)s, nd et (n+1)p du cation métallique. Leur configuration géométrique dépend du nombre de ligands et du type de recouvrement des orbitales originelles. La configuration la plus stable est obtenue avec 6 ligands en recouvrement sigma de leurs orbitales avec celles du cation métallique. (Un recouvrement sigma est un recouvrement axial. On dit dans ce cas des ligands qu’ils sont sigma-donneurs.) Elle conduit à un octaèdre dont les sommets sont occupés par les ligands, le cation étant au centre. Elle comporte 15 OM auxquelles on a donné les noms suivants :
a1g et a*1g pour les combinaisons avec l’orbitale (n+1)s,
t1u et t*1u (triplement dégénérées) pour les combinaisons avec les orbitales (n+1)p,
eg et e*g (doublement dégénérées) et t2g (triplement dégénérée) pour les combinaisons avec les orbitales nd.
Les orbitales notées avec un astérisque sont anti-liantes (les électrons de cette orbitale se repoussent). Elles ont d'ailleurs une énergie supérieure à celle des orbitales d’origine des ligands. Elles ne donnent pas lieu à une liaison de coordination. Les orbitales t2g ont leur lobe qui n'est pas orienté dans une direction convenable pour assurer un recouvrement sigma. Elles sont non liantes. Il reste donc 6 orbitales liantes possibles pour former un complexe dès lors qu'elles ont une énergie inférieure à celle des orbitales donneuses de doublet des ligands : une orbitale a1g, deux orbitales eg et trois orbitales t1u. Les 3 orbitales non liantes t2g (dont l'énergie n'est pas modifiée) sont occupées en priorité par les électrons appartenant au cation métallique.
Remarque 1 : cette notation fait référence à la théorie des groupes de symétrie octaédrique. Pour qu’il y ait recouvrement entre orbitale du métal et orbitale d’un ligand, il faut qu’elles présentent les mêmes symétries.
Remarque 2 : si l’on s’en tient au raisonnement qui précède, les orbitales a1g, eg et t1u devraient avoir une forme d’onde différente. Une orbitale s n’a pas la même forme qu’une orbitale p. En fait, pour des raisons de symétrie, il y a hybridation de ces orbitales. Ainsi par exemple, au lieu d’avoir une orbitale s et trois orbitales p, on a quatre orbitales sp.
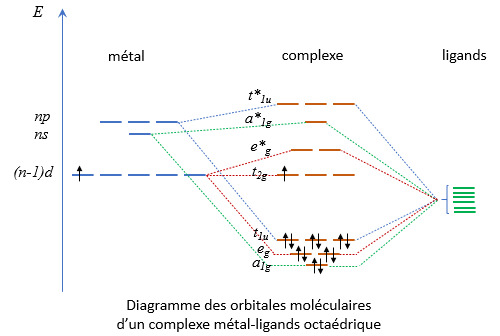
Le titane (III) hexahydraté [Ti(H2O)6]3+, dont le nom officiel est hexaaquo titane (III), illustre ce schéma (voir la figure ci-dessus). L’atome de titane possède 22 électrons (1s2 ; 2s2, 2p6 ; 3s2, 3p6 ; 4s2, 3d2). La configuration électronique du cation Ti3+ est (1s2 ; 2s2, 2p6 ; 3s2, 3p6 ; 3d1). L'électron de valence (3d1) de l'ion Ti3+ reste sur l'une des orbitales t2g alors que les doublets non liants des molécules H2O occupent les orbitales a1g, eg et t1u pour former le complexe [Ti(H2O)6]3+.
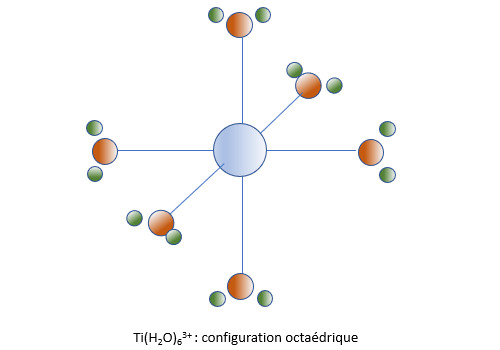
Idem pour l'ion hexacyanoferrate [Fe(CN)6]4-, aussi appelé ferrocyanure. L'ion fer (II) a pour configuration électronique (1s2 ; 2s2, 2p6 ; 3s2, 3p6 ; 4s2, 3d4). Les 6 électrons de sa bande de valence occupent les 3 orbitales t2g du complexe et les doublets non liants des ions cyanures CN- occupent les 6 orbitales liantes a1g, eg et t1u. Le ferrocyanure entre dans la composition du bleu de Prusse, un pigment connu de longue date.
Les ligands
Si l’on se reporte à la présentation de la liaison de coordination ci-dessus, on constatera qu’il y a une grande similarité avec la notion de réaction acide/base de Lewis. Une base de Lewis est une molécule ou un ion capable de donner un doublet d’électron et un acide de Lewis une molécule ou un ion capable de le recevoir. De fait, dans la liste des ligands les plus courants, on retrouvera un grand nombre de bases de Lewis.
Parmi les molécules simples, on peut citer par exemple la molécule H2O et la molécule CO. Dans la molécule H2O, l’atome d’oxygène conserve deux doublets électroniques non liants dans sa bande de valence, ce qui en fait un ligand qui intervient dans de nombreux complexes. La molécule CO quant à elle représente un cas intéressant. La liaison entre carbone et oxygène est triple. Elle est en fait constituée de deux liaisons covalentes standard et d’une liaison de coordination, liaison dans laquelle l’atome d’oxygène joue le rôle de donneur et l’atome de carbone celui d’accepteur. (Ce n’est cependant pas un complexe : il n’y a pas d’atome ou de cation central.) On peut donc considérer que la bande de valence de l’atome de carbone comporte 4 doublets : deux doublets covalents, un doublet de coordination « prêté » par l’atome d’oxygène, et un doublet non liant. Idem pour l’oxygène… qui s’est dépouillé d’un doublet au profit du carbone. La molécule CO comporte donc elle aussi deux doublets non-liants, mais cette fois ils ne sont pas portés par le même atome. L’atome de carbone en porte un et l’atome d’oxygène porte l’autre.
Parmi les ligands inorganiques, on trouve aussi de nombreux ions ou radicaux simples. L’ion cyanure CN- et l’ion hydroxyle OH- en sont des exemples que l’on peut comprendre facilement à partir de ce que nous venons de dire. C’est évident pour l’ion hydroxyle (l’atome d’oxygène possède trois doublets non liants si l’on tient compte de la charge électrique de l’ion). Pour l’ion cyanure, on peut faire un raisonnement par analogie avec la molécule CO. En prenant en compte la charge de l’ion, on a une répartition similaire des doublets entre atome de carbone de carbone et atome d’azote.
Les ions NO2- et NH2- ainsi que l’ammoniac NH3 rentrent également dans la composition de nombreux complexes. C’est chaque fois l’atome d’azote qui est porteur du doublet non liant. Même chose pour les phosphines, phosphinites et phosphonites, avec cette fois l’atome de phosphore qui est porteur du doublet.
Dans ce tour d’horizon des ligands inorganiques, il convient de citer deux cas particuliers importants : celui des ions halogénures et celui de l’ion hydrure. Les halogénures sont connus pour entrer dans la composition de nombreux sels dans lesquels ils entretiennent des liaisons ioniques. Du fait de la constitution de leur bande de valence, les ions halogénures se révèlent également être des ligands forts. La charge électronique qu’ils portent leur permet en effet d’avoir quatre doublets non liants ! Pour ce qui est de l’ion hydrure H-, il est facile de comprendre qu’il engage facilement son doublet d’électrons dans une liaison avec un élément plus électronégatif que lui (ce qui est le cas de tous les cations métalliques).
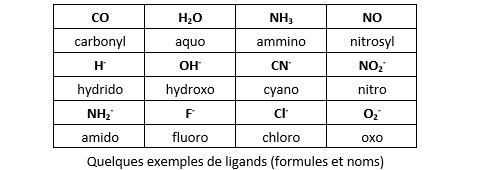
Remarque : on peut classer empiriquement les ligands en fonction de leur énergie de liaison :
S2− < Cl− < F− < O2− < H2O < NH3 < NO− < CN-< CO
Le ligand carbonyle CO est clairement le plus fort et comme on le voit le ligand H2O > a une force moyenne.
Certaines molécules organiques se prêtent à des liaisons de coordination qui peuvent donner lieu à la constitution de complexes. Nous reviendrons sur ce sujet dans le post suivant.
La nomenclature des complexes suit une règle de nommage simple. Lorsqu’il s’agit d’un anion la règle est la suivante :
[nom des ligands] [nom du métal] -ate (degré d’oxydation)
Lorsqu’il s’agit d’un composé neutre ou d’un cation, la règle est :
[nom des ligands] [nom du métal] (degré d’oxydation)
Les ligands sont généralement cités dans l’ordre alphabétique. Exemples :
[Fe(CN)6]4- hexacyanoferrate (II)
[Ti(H2O)6]3+ hexaaquo titane (III)
[PtCl(NH3)5]3+ chloro pentaammino platine (IV)
Géométrie et nombre de coordination
Nous avons jusqu’à présent cité en exemple des complexes comportant 6 ligands. C’est la configuration la plus répandue mais il en existe d’autres en fonction du nombre de ligands :
linéaire avec 2 ligands comme le diamine cuivre [Cu(NH3)2]+,
tétraédrique avec 4 ligands comme le tétracarbonyle de nickel Ni(CO)4,
plane carrée avec 4 ligands comme le tétracyanure de nickel [Ni(CN)4]2-,
bipyramide trigonale avec 5 ligands comme l’hydrocarbonyle de cobalt hydrocarbonyle de cobalt HCo(CO)4 ,
etc...
On appelle nombre de coordination le nombre d’atomes auquel le cation central est lié.

Règle des 18 électrons
La règle des 18 électrons stipule que les complexes métalliques les plus stables sont obtenus lorsque la couche de valence des cations au centre du complexe est saturée. Pour un métal de transition, cette couche peut accueillir 18 électrons, d’où le nom de cette règle.
Cette règle des 18 électrons est loin de s’appliquer de manière systématique. Il semble que la stabilité des complexes soit plutôt liée à des considérations de symétrie, les formes géométriques comme celles indiquées ci-dessus étant privilégiées, que la règle soit respectée ou pas. Quelques exemples vont nous permettre d’illustrer notre propos.
Configuration tétraèdrique
Le tétracarbonyle de nickel Ni(CO)4 est l’exemple typique de complexe tétraédrique dont la bande de valence est remplie. Le tétrachlorure de titane (IV) TiCl4, par contre, est loin de faire le plein. Le cation central Ti4+ dont la configuration électronique est (4s0, 3d0) n’accueille que 4 paires d’électrons, soient 8 électrons au total sur les 18 possibles dans sa bande de valence. Le tétrachlorure de vanadium (IV) VCl4 présente lui aussi un fort déficit en électrons de la bande de valence du vanadium (9 électrons au lieu de 18). Même chose pour le dichlorure de titanocène (C2H5)2TiCl2. Le titanocène est un métallocène, un complexe dans lequel le cation est relié à deux ligands cyclopentadiényles par une liaison éta (les anions C5H5 sont aromatiques).
Nota : les composés complexes dont les ligands sont des ions chlorure ou fluorure n’ont le plus souvent pas besoin de la règle des 18 pour être stables.
Plan carré
Dans le tétracyanure de nickel (II) [Ni(CN)4]2- la configuration de l’ion Ni2+ est (3d8). Les ions cyanure qui forment un carré autour de lui ne lui apportent que 3 autres paires d’électrons et le compte n’y est pas... Dans le cis-diammino dichloro platine (II) Pt Cl2(NH3)2 le platine, qui est oxydé deux fois, conserve également une orbitale libre. Le cis-diammino dichloro platine est aussi appelé CDDP ou cisplatine. C’est un médicament anticancéreux.
Nota : le préfixe cis- indique que les ligands chloro sont adjacents. Dans le cas contraire, on utilise le préfixe trans-.
Bipyramide trigonale
L’hydrocarbonyle de cobalt HCo(CO)4 a une configuration bipyramide trigonale. Le cobalt a la configuration électronique (4s2, 3d7). Il fait le plein avec les quatre doublets apportés par les groupes CO et l’électron célibataire de l’atome d’hydrogène. Idem pour l’octacarbonyle de dicobalt Co2(CO)8 dans lequel il se décompose à l’ambiante et qui forme un empilage de deux bipyramides trigonales.
Octaèdre
Les composés octaédriques qui satisfont à la règle des 18 sont nombreux. L’hexaaquo fer (II) [Fe(H2O)6]2+ la respecte mais pas l’hexaaquo titane [Ti(H2O)6]3+. Dans le composé HMn(CO)5 le manganèse fait le plein, de même que dans le composé le décacarbonyle de dimanganèse Mn2(CO)10 composé d’un empilage de deux octaèdres.
Dans l’hexa-amine chrome (III) [Cr(NH3)6]3+ le chrome conserve trois électrons célibataires (15 électrons au lieu de 18). Dans l’hexacarbonyle de vanadium V(CO6), le vanadium (4s2, 3d3) accueille quant à lui 6 doublets non liants, soient 17 électrons au lieu de 18.
Le chlorure de molybdène (V) (MoCl5)2 est constitué de deux octaèdres qui partagent deux ions chlorure de leur base carré. La configuration électronique du cation molybdène (V) est (5s1). Dans le chlorure de molybdène, sa bande de valence n’accueille donc que 4 doublets et deux électrons non appariés supplémentaires. Il ne fait pas le plein.

Dans le réactif de Schweizer [Cu(NH3)4(H2O)2]2+ l’ion Cu2+ possède 9 électrons dans sa bande de valence. Il se trouve donc largement en excès par rapport à la règle des 18 électrons ! L’explication réside dans le fait que les liaisons autour du cuivre ont une forte composante ionique.
Le composé hexa-aquo fer (III) [Fe(H2O)6]3+ est un cas tout à fait particulier. Son caractère nettement paramagnétique incite à penser qu’il conserve les 5 électrons célibataires de la sous-couche 3d de l’ion fer (III) (voir plus bas les propriétés magnétiques des complexes). Ceci ne peut s’expliquer que si deux orbitales 4d sont mobilisées par le composé. L’octaèdre serait alors constitué par l’hybridation de l’orbitale 4s, des trois orbitales 4p et de ces deux orbitales 4d (hybridation sp3d2).
Complexes multi-métalliques
Nous avons jusqu’à présent supposé que le corps central était un atome ou un cation métallique. Les atomes métalliques se combinent facilement entre eux et il existe des complexes polyatomiques dont le noyau central est un polymère Mn (M étant un atome métallique). C’est le cas par exemple du dodécarbonyle trifer Fe3(CO)12. Le noyau central est constitué par trois atomes de fer formant un triangle. Chaque atome est porteur de quatre liaisons de coordination avec des ligands carbonyles CO. La configuration du fer reste du type octaédrique (6 liaisons). Dans le dodécarbonyle tétracobalt Co4(CO)12, les quatre atomes de cobalt occupent le sommet d’un tétraèdre et portent chacun trois liaisons de coordination avec des ligands carbonyles.
Couleur et propriétés magnétiques
Les composés complexes ont une couleur franche (certains sont utilisés comme pigment) et des propriétés magnétiques bien définies. Les différents niveaux d’énergie des orbitales permettent d’expliquer ces propriétés (on utilise dans ce cas la théorie des champs cristallins qui est une adaptation de la théorie des OA au cas d’un réseau). La couleur d’un complexe est la couleur complémentaire de la fréquence absorbée lors d’un saut d’un niveau à un autre. Les propriétés magnétiques quant à elles s’expliquent par la différence d’énergie entre les différentes orbitales.
Les électrons d’une même couche ont tendance à se répartir sur les différentes orbitales de façon à minimiser le nombre d’électrons appariés (règle de Hund). Cela résulte du fait que l’appariement des électrons (c.à.d le fait deux électrons de spin opposé occupent la même orbitale) a un coût énergétique. Dans la figure représentant l’énergie des orbitales moléculaires nous avons supposé qu’il y avait un appariement maximal (règle de Hund non respectée). Ceci dépend en fait de l’écart d’énergie entre les orbitales. Si le coût énergétique de l’appariement des électrons est supérieur à cet écart, les électrons vont avoir tendance à se répartir entre les deux niveaux pour minimiser leur énergie totale. La règle de Hund sera alors respectée. Dans le cas contraire (comme dans la figure) ils vont s’apparier. On caractérise ces deux situations en disant que la première est de type champ faible – spin fort et la seconde champ fort - spin faible. Cela a une incidence directe sur les propriétés magnétiques. Un complexe champ faible – spin fort est paramagnétique alors qu’un complexe champ fort – spin faible est diamagnétique. Question de spin ! On peut d’ailleurs remarquer qu’un complexe champ fort – spin faible est souvent incolore. Le delta d’énergie pour faire passer un électron sur un niveau orbitalaire d’énergie supérieure est souvent trop élevé pour être dans le domaine visible.
Pour en savoir plus :
post d’introduction à la chimie
post sur les électrons
post sur les électrons dans les liaisons covalentes
post sur les liaisons chimiques
post sur les orbitales moléculaires
post sur la valence
post sur la géométrie des molécules
post sur la cohésion de la matière
post sur la classification périodique des éléments
post sur les métaux de transition
post sur les acides et bases
post sur les complexes et les ligands : l’approche covalente
post sur les complexes et les ligands : exemples
post sur le phosphore
post sur l’azote
post sur les matériaux magnétiques
index
2 notes
·
View notes
Text
La théorie des bandes et les semi-conducteurs
Comme on l’a vu dans le post consacré aux électrons d’un atome, ceux-ci ne peuvent occuper que des niveaux d’énergie bien définis. Lorsque les atomes sont regroupés dans un solide, on pourrait s’attendre à retrouver exactement la même situation. C’est compter sans le principe d’exclusion de Pauli. Si les électrons de deux atomes voisins avaient le même niveau d’énergie, il pourrait exister une superposition de deux états de niveau d’énergie identique s’étendant sur ces deux atomes et que pourrait occuper simultanément deux électrons. Or la physique quantique l’interdit. Pour contourner l’interdiction, il se produit une démultiplication des états d’énergie possibles. La démultiplication des états à partir d’un même niveau d’énergie initial conduit à la formation d’une bande d’énergie. Et comme le nombre d’atomes d’un solide est astronomique, la densité d’états électroniques au sein d’une bande est telle qu’on peut pratiquement considérer que tous les niveaux d’énergie sont permis au sein de celle-ci. La répartition de ces bandes dépend de la nature du solide considéré : toutes les configurations sont possibles. Deux bandes successives peuvent se chevaucher ou être séparées. L’écart entre deux bandes est appelé le gap. Ce gap peut être relativement faible ou élevé.
Deux bandes jouent un rôle particulier :
La dernière bande entièrement occupée par des électrons est appelé bande de valence. Les électrons de cette bande participent à la cohésion du solide. Ils sont plus ou moins piégés dans des puits de potentiel.
La bande d’énergie immédiatement supérieure peut être vide ou partiellement occupée. On l’appelle bande de conduction car ce sont les électrons qui l’occupent qui permettent (le cas échéant) la conduction électrique.
On caractérise cette situation avec une grandeur qui est une propriété du solide considéré : le niveau de Fermi. Le niveau de Fermi est le niveau d'énergie le plus élevé occupé par un électron si on fait l’hypothèse que le solide est au zéro absolu. La position du niveau de Fermi par rapport aux bandes détermine les propriétés électriques d’un solide :
Premier cas : la bande de conduction est vide et séparée par un gap important de la bande de valence (6 eV et plus). Les électrons restent piégés. Le solide est isolant.
Deuxième cas : la bande de conduction est partiellement occupée (ou la bande de valence et la bande de conduction se chevauchent). Un champ électrique de faible intensité peut faire passer un électron à un niveau d'énergie qui lui permet de circuler dans le solide. Celui-ci est conducteur.
Troisième cas : la bande de conduction est vide mais le gap est relativement faible (moins de 2,5 eV). Le solide est donc isolant à température nulle, mais le passage des électrons de la bande de valence à la bande de conduction est possible (en chauffant le solide ou par un autre biais comme on le verra un peu plus loin). Le solide est un semi-conducteur.

Remarque : en toute rigueur, on devrait préciser que ceci n’est valable qu’au zéro absolu car l’agitation thermique conduit à exciter les électrons qui peuvent, le cas échéant, changer de bande.
Semi-conducteurs
Dans un semi-conducteur, comme on l’a vu plus haut, la largeur de la bande interdite entre bande de valence et bande de conduction est faible (1,12 eV pour le silicium, 0,66 eV pour le germanium). Dès lors qu’on apporte cette énergie aux électrons, ceux-ci peuvent passer de la bande de valence à la bande de conduction et circuler dans le matériau.
Cette énergie peut être apportée en chauffant le solide mais ça ne présente en pratique pas grand intérêt… Une autre manière consiste à illuminer le solide : les applications sont beaucoup plus nombreuses. Les composants que l’on utilise de cette façon sont appelés composants optoélectroniques ou, de façon plus condensée, optroniques.
Il existe une troisième façon de permettre la circulation, sous certaines conditions, de courant dans les semi-conducteurs, c’est le dopage. Le dopage d’un semi-conducteur consiste à remplacer certains atomes du réseau cristallin par d’autres atomes ayant un nombre d’électrons de valence différents.
Prenons le cas du silicium qui est le matériau le plus utilisé pour réaliser des composants électroniques. Il possède 4 électrons de valence, tous quatre engagés dans une liaison covalente avec ses voisins. Si on remplace un certains nombre d’atomes de silicium du réseau cristallin par des atomes de phosphore, on introduit dans ce réseau des atomes ayant 5 électrons dans leur couche externe. Ce cinquième électron, qui n'est engagé dans aucune liaison covalente, peut être facilement excité et passer dans la bande de conduction où il se comportera comme un électron libre. Ce type de dopage est appelé dopage N.
Si au lieu d’introduire dans le silicium des atomes pentavalents on y insère des atomes trivalents (comme le bore) on va se retrouver dans une situation où il y a des « trous » dans la structure électronique du réseau. Ce trou peut être comblé par un électron en provenance d’un atome voisin, donc se déplacer. Dans ce type de semi-conducteur, ce ne sont plus les électrons mais les trous qui sont susceptibles de conduire le courant. On parle dans ce cas de dopage P.
Jonction PN.
Une jonction PN est réalisée en dopant de manière différente deux zones adjacentes du semi-conducteur. La jonction est constituée par l’interface entre ces deux zones. Si on applique une différence de potentiel positive entre la zone P et la zone N, trous et électrons sont attirés vers la jonction. Dans un premier temps, électrons et trous se recombinent, formant ainsi une micro-zone chargée électriquement autour de la jonction (on parle de capacité de jonction). Puis un courant s’établit, les électrons circulant dans la bande de conduction vers le pôle positif et les trous se déplaçant dans la zone de valence vers le pôle négatif. Si on applique une différence de potentiel de signe opposé, trous et électrons s’éloignent de la jonction et aucun courant électrique ne peut circuler au travers de la jonction. Ce type de composant qui n’est passant que dans un seul sens est ce que l’on appelle une diode.
Remarque : lorsque la recombinaison électron-trou se traduit par l’émission d’un photon en lumière visible, cette diode est appelée électroluminescente (LED).
Pour illustrer l’effet de la polarisation sur une diode, on utilise la notion de barrière de potentiel. Le gap entre bande de conduction et bande de valence établit une barrière de potentiel qui entrave la mobilité des électrons (ou les trous). En appliquant une polarisation positive aux bornes de la diode, on établit un champ électrique dans le semi-conducteur qui donne aux électrons libres et aux trous l’énergie potentielle nécessaire pour se déplacer dans tout le semi-conducteur. On dit de cette polarisation qu’elle permet d’abaisser la barrière de potentiel. A l’inverse, si on applique une polarisation négative, celle-ci va renforcer la barrière de potentiel puisqu’elle augmente l’énergie qui serait nécessaire pour que es électrons ou les trous franchissent la jonction.

Transistor bipolaire
Un transistor bipolaire est constitué d’un empilage de deux jonctions : NPN ou PNP. Prenons le cas d’un transistor NPN. Les deux zones N sont appelées respectivement collecteur et émetteur. La zone P est la base. L’épaisseur de la base est faible : de l’ordre d’un micron, voire moins. Supposons que l’on applique une différence de potentiel de plusieurs volts (voire plusieurs dizaines ou plusieurs centaines de volts) entre le collecteur et l’émetteur (oui, je sais, ça paraît bizarre d’appeler collecteur la zone reliée au pôle positif et émetteur celle reliée au pôle négatif, mais on se place du côté des électrons). Supposons maintenant que l’on applique une tension positive (0,6 V) sur la base. A priori, la jonction base-émetteur est passante et la jonction collecteur-base est bloquée. Mais l’épaisseur de la base est très fine. Dès lors que la capacité de jonction est chargée, les électrons injectés par l’émetteur dans la base la traversent sans se recombiner et pénètrent dans le collecteur. Une fois dans le collecteur, ces électrons se retrouvent dans la bande de conduction de celui-ci et se comportent comme des électrons libres. (Pour reprendre l’image de la barrière de potentiel, on peut dire que la polarisation de la jonction base-émetteur donne aux électrons le potentiel nécessaire pour franchir la jonction collecteur-émetteur bien que celle-ci soit polarisée en inverse.) Le transistor est donc passant. L’intensité du courant qui le traverse est contrôlée par celle du courant de base.

L’effet transistor a été découvert en 1947 par John Bardeen, William Shockley et Walter Brattain. Cette découverte est à la base d’une révolution qui a changé le cours de notre civilisation : la révolution électronique !

Nota : on a, depuis, imaginé d’autres configurations permettant de réalises des transistors : transistors à effet de champ (FET et JFET), transistor de type métal-oxyde-semi-conducteur (MOS et CMOS). Mais ça, s’est une autre histoire…
Pour en savoir plus :
post sur les électrons
post sur les électrons dans les atomes
post d’introduction à la mécanique quantique
post sur les liaisons chimiques
post sur le silicium
post sur la cohésion de la matière
post sur la classification périodique des éléments
index
3 notes
·
View notes
Text
Gènes et ADN
La génétique est la branche de la biologie qui s’intéresse aux gènes et à l’hérédité. De nos jours la génétique est principalement centrée sur l’étude de l’ADN (acide désoxyribonucléique) et de l’ARN (acide ribonucléique), son rôle dans le fonctionnement des cellules et dans l’expression des traits héréditaires (les phénotypes) et sa réplication.
L’ADN a été isolé pour la première fois en 1869 par un biologiste suisse, Friedrich Miescher. A la même époque, Gregor Mendel énonçait les règles de l’hérédité mais on ne fit pas tout de suite le lien entre ADN et hérédité. C’est à la fin du XIXème siècle qu’on découvre les chromosomes et leur rôle dans la transmission de l’hérédité. La théorie chromosomique de l’hérédité est proposée en 1902 par Walter Sutton. Il montre qu’elle est compatible avec les règles énoncées par Mendel et prédit l’existence de facteurs biologiques de l’hérédité. Les termes de gènes, génotype et phénotype apparaissent peu de temps après dans la littérature scientifique. Thomas Morgan démontre en 1911 que les chromosomes sont les supports des gènes que la théorie de Sutton prédisait. Il publie en 1913 la carte génétique du chromosome X de la mouche du vinaigre (dont le nom scientifique est drosophile).
L’analyse plus précise du rôle et de la structure des chromosomes et la mise en évidence du rôle central joué par l’ADN est le fruit de longues années de recherche. C’est en 1944 qu’ Oswald Avery, Colin MacLeod, et Maclyn McCarty démontrent le rôle de l’ADN comme support de l’hérédité. La structure en double hélice de l’ADN est découverte en 1953 par Francis Crick et James Watson et en 1955 on découvre l’ARN polymérase, une enzyme qui permet la réplication de l’ADN.Dans les années 1960, les travaux de François Jacob et Jacques Monod permettent de mieux comprendre le séquencement des gènes dans l’ADN et établissent la différence entre gènes structuraux et gènes régulateurs. La notion de code génétique commence à se diffuser dans la communauté scientifique.
Dans le même temps, Barbara McClintok découvre le mécanisme de transposition dont les implications sur les gènes commencent seulement à être élucidés.
Les premiers travaux de manipulation génétique datent des années 1980 mais ce n’est qu’à la fin du XXème siècle que la technologie permet la cartographie du génome humain : cartographie d’un chromosome en 1999 et séquençage complet du génome en 2003. La technologie permet aujourd’hui le séquençage d’une molécule d’ADN en quelques heures seulement.
Génome, ADN et ARN
Le génome est l’ensemble des informations génétiques caractérisant une espèce. Le support de ces informations génétiques est constitué de molécules ADN (à l’exception de certains virus pour lesquels cette information est codé sur une molécule d’ARN). Les molécule d’ADN et d’ARN sont des chaînes polymères constituées denucléotides. Un nucléotide est une molécule composée d’unpentose sous forme cyclique (désoxyribose C5H10O4 ou ribose C5H10O5), d’une base nucléique (aussi appelé base azotée) et de un à trois groupes phosphate. Le désoxyribose est dérivé du ribose par perte d’un atome

Remarque : dans la figure qui précède, on a utilisé la représentation topologique des molécules organiques. Dans cette représentation, lorsque aucun atome n’est spécifié à l’extrémité d’un segment c’est qu’il s’agit d’un atome de carbone. Les atomes d’hydrogène reliés à un atome de carbone non mentionné explicitement ne sont pas non plus représentés. Par exemple, dans le schéma représentant le désoxyribose, il y a quatre intersections muettes. Il y a donc un atome de carbone à chacune d’entre elles et cinq au total. Chaque atome de carbone portant quatre liaisons covalentes, il y a donc 5 atomes d’hydrogène non représentés, un sur le carbone 1, deux sur le carbone 2 et un sur les carbones 3 et 4. La numérotation se fait à partir de l’atome d’oxygène, l’atome 5 étant celui qui n’est pas dans le cycle. (En toute logique, on aurait d’ailleurs pu ne pas représenter le carbone 5 et les deux atomes d’hydrogène qui lui sont rattachés.) Cette représentation est également utilisée par la suite.
Le chaînage des nucléotides au sein d’une molécule d’ADN (ou d’ARN) se fait par l’intermédiaire d’une liaison phosphodiester entre le cinquième carbone du pentose (celui qui est hors du cycle) et le troisième
Une liaison phosphodiester lie un groupe phosphate à deux molécules organiques pour former une molécule dont la formule générique peut s’écrire R-O-(PO2)-O-CH2-R’. Ces liaisons sont des liaisons ester : elle passe par un atome d’oxygène qui est relié à un élément déjà porteur d’une liaison double avec un autre atome d’oxygène : R-(X=O)-O-R’.

Nous verrons dans des posts ultérieurs comment l’ADN des cellules eucaryote se répartit entre chromosomes, ce qu’est un gène et comment il est codé sous forme de bases nucléiques.
Pour en savoir plus :
post sur les cellules eucaryotes et les chromosomes
post sur les bases nucléiques
post sur les gènes et les codons
l’ADN en bref
post sur les couples ADP/ATP et NAD/NADH
post sur la photosynthèse et la respiration cellulaire
glossaire de biologie moléculaire
post d’introduction à la chimie du vivant
post sur les éléments chimiques
post sur les molécules des organismes vivants
post sur l’abondance des éléments dans le corps humain
post sur les glucides
post sur les hydrocarbures aromatiques
glossaire de chimie organique
index
#chimie#chimie organique#biologie#adn#gène#nucléotide#protéine#arn#codon#adénine#thymine#cytosine#guanine#uracile#ribonucléique#désoxyribonucléique#acide désoxyribonucléique#acide aminé#chromosome
2 notes
·
View notes