#Microbial taxonomic
Explore tagged Tumblr posts
Visit Tumblr Blog
Explore Tumblr blogs with no restrictions, modern design and the best experience.
Last Seen Tumblr Blogs





Fun Fact
25% of US internet users with an annual income of $80-100K use Tumblr.
Text
Also preserved in our archive
By Vijay Kumar Malesu
In a recent pre-print study posted to bioRxiv*, a team of researchers investigated the predictive role of gut microbiome composition during acute Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) infection in the development of Long Coronavirus Disease (Long COVID) (LC) and its association with clinical variables and symptom clusters.
Background LC affects 10–30% of non-hospitalized individuals infected with SARS-CoV-2, leading to significant morbidity, workforce loss, and an economic impact of $3.7 trillion in the United States (U.S.).
Symptoms span cardiovascular, gastrointestinal, cognitive, and neurological issues, resembling myalgic encephalomyelitis and other post-infectious syndromes. Proposed mechanisms include immune dysregulation, neuroinflammation, viral persistence, and coagulation abnormalities, with emerging evidence implicating the gut microbiome in LC pathogenesis.
Current studies focus on hospitalized patients, limiting generalizability to milder cases. Further research is needed to explore microbiome-driven predictors in outpatient populations, enabling targeted diagnostics and therapies for LC’s heterogeneous and complex presentation.
About the study The study was approved by the Mayo Clinic Institutional Review Board and recruited adults aged 18 years or older who underwent SARS-CoV-2 testing at Mayo Clinic locations in Minnesota, Florida, and Arizona from October 2020 to September 2021. Participants were identified through electronic health record (EHR) reviews filtered by SARS-CoV-2 testing schedules.
Eligible individuals were contacted via email, and informed consent was obtained. Of the 1,061 participants initially recruited, 242 were excluded due to incomplete data, failed sequencing, or other issues. The final cohort included 799 participants (380 SARS-CoV-2-positive and 419 SARS-CoV-2-negative), providing 947 stool samples.
Stool samples were collected at two-time points: weeks 0–2 and weeks 3–5 after testing. Samples were shipped in frozen gel packs via overnight courier and stored at −80°C for downstream analyses. Microbial deoxyribonucleic acid (DNA) was extracted using Qiagen kits, and metagenomic sequencing was performed targeting 8 million reads per sample.
Taxonomic profiling was conducted using Kraken2, and functional profiling was performed using the Human Microbiome Project Unified Metabolic Analysis Network (HUMAnN3).
Stool calprotectin levels were measured using enzyme-linked immunosorbent assay (ELISA), and SARS-CoV-2 ribonucleic acid (RNA) was detected using reverse transcription-quantitative polymerase chain reaction (RT-qPCR).
Clinical data, including demographics, comorbidities, medications, and symptom persistence, were extracted from EHRs.
Machine learning models incorporating microbiome and clinical data were utilized to predict LC and to identify symptom clusters, providing valuable insights into the heterogeneity of the condition.
Study results The study analyzed 947 stool samples collected from 799 participants, including 380 SARS-CoV-2-positive individuals and 419 negative controls. Of the SARS-CoV-2-positive group, 80 patients developed LC during a one-year follow-up period.
Participants were categorized into three groups for analysis: LC, non-LC (SARS-CoV-2-positive without LC), and SARS-CoV-2-negative. Baseline characteristics revealed significant differences between these groups. LC participants were predominantly female and had more baseline comorbidities compared to non-LC participants.
The SARS-CoV-2-negative group was older, with higher antibiotic use and vaccination rates. These variables were adjusted for in subsequent analyses.
During acute infection, gut microbiome diversity differed significantly between groups. Alpha diversity was lower in SARS-CoV-2-positive participants (LC and non-LC) than in SARS-CoV-2-negative participants.
Beta diversity analyses revealed distinct microbial compositions among the groups, with LC patients exhibiting unique microbiome profiles during acute infection.
Specific bacterial taxa, including Faecalimonas and Blautia, were enriched in LC patients, while other taxa were predominant in non-LC and negative participants. These findings indicate that gut microbiome composition during acute infection is a potential predictor for LC.
Temporal analysis of gut microbiome changes between the acute and post-acute phases revealed significant individual variability but no cohort-level differences, suggesting that temporal changes do not contribute to LC development.
However, machine learning models demonstrated that microbiome data during acute infection, when combined with clinical variables, predicted LC with high accuracy. Microbial predictors, including species from the Lachnospiraceae family, significantly influenced model performance.
Symptom analysis revealed that LC encompasses heterogeneous clinical presentations. Fatigue was the most prevalent symptom, followed by dyspnea and cough.
Cluster analysis identified four LC subphenotypes based on symptom co-occurrence: gastrointestinal and sensory, musculoskeletal and neuropsychiatric, cardiopulmonary, and fatigue-only.
Each cluster exhibited unique microbial associations, with the gastrointestinal and sensory clusters showing the most pronounced microbial alterations. Notably, taxa such as those from Lachnospiraceae and Erysipelotrichaceae families were significantly enriched in this cluster.
Conclusions To summarize, this study demonstrated that SARS-CoV-2-positive individuals who later developed LC exhibited distinct gut microbiome profiles during acute infection. While prior research has linked the gut microbiome to COVID-19 outcomes, few studies have explored its predictive potential for LC, particularly in outpatient cohorts.
Using machine learning models, including artificial neural networks and logistic regression, this study found that microbiome data alone predicted LC more accurately than clinical variables, such as disease severity, sex, and vaccination status.
Key microbial contributors included species from the Lachnospiraceae family, such as Eubacterium and Agathobacter, and Prevotella spp. These findings highlight the gut microbiome’s potential as a diagnostic tool for identifying LC risk, enabling personalized interventions.
*Important notice: bioRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
Journal reference: Preliminary scientific report. Isin Y. Comba, Ruben A. T. Mars, Lu Yang, et al. (2024) Gut Microbiome Signatures During Acute Infection Predict Long COVID, bioRxiv. doi:https://doi.org/10.1101/2024.12.10.626852. www.biorxiv.org/content/10.1101/2024.12.10.626852v1.full
#mask up#public health#wear a mask#pandemic#wear a respirator#covid#still coviding#covid 19#coronavirus#sars cov 2#long covid#AI
36 notes
·
View notes
Text
lil guys of the Ediacaran
the Ediacaran period was around ~635 million years ago, happening before the Cambrian. This time saw the rise of the first group of animals and the oldest multicellular life. The fossil record is sparse due to the fact that the creatures did not have hard shells, which are the thing most easily fossilised.
the Ediacaran biota appeared ~570 million years ago, and are very taxonomically ambiguous, they may relate to some groups, like cnidaria and protozoans, but some have suggested completely new phylums that we do not have today.
the majority of Ediacaran biota were sessile (lacking the ability to move by themselves) and were soft-bodied. many also lacked mouths and a gut.
here are some lil guys!

These frondose fossils are very controversial, considering they appear to be leafy plants, but may very well be anything from animal or protist, to stem fungi (https://www.science.org/doi/10.1126/science.1099727)
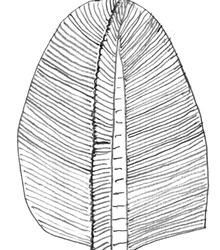
this is an Erniettamorph and we know basically nothing about these! The fossils were found in places where photosynthesis would not have been possible, and one type of these fossils has been found with the tubes completely covered and filled with sand, which makes the possibility of osmotic feeding from the surrounding water difficult as it would reduce the metabolically active volume. (https://www.pnas.org/doi/full/10.1073/pnas.0904836106)

Dickinsonia is a more studied animal. and it probably was an animal, due to cholesterol molecules found in fossils. Fossils range from millimetres to ~1.4 metres, and have near bilateral symmetry and rib-like segments. They were a mobile creature and ate microbial mats (trace fossils have been found which are most likely impressions from feeding) (https://doi.org/10.1017/S175569102300004X) A study from 2022 has suggested that they temporarily stuck themselves to substrate with mucus, which suggests a life in shallow waters (https://doi.org/10.1017/S0016756821000194)
support me on ko-fi?
#some of these articles are not freely available#so i might get the pdfs for people and drop them in a google drive if people would like#marine biology#ediacaran period#eddie in the ocean#dickinsonia#erniettamorph#frondose#fossils#paleontology#im on a paleo posting spree because this is what we r doing in lectures#i think ive had one singular lecture about modern animals (it was a cephalopods one)#oh wait no i had one on cnidaria and porifera ignore me#queue are in the ocean
49 notes
·
View notes
Text
Data-Independent Acquisition Mass Spectrometry as a Tool for Metaproteomics: Interlaboratory Comparison Using a Model Microbiome
Mass spectrometry (MS)-based metaproteomics is used to identify and quantify proteins in microbiome samples, with the frequently used methodology being Data-Dependent Acquisition mass spectrometry (DDA-MS). However, DDA-MS is limited in its ability to reproducibly identify and quantify lower abundant peptides and proteins. To address DDA-MS deficiencies, proteomics researchers have started using Data-Independent Acquisition Mass Spectrometry (DIA-MS) for reproducible detection and quantification of peptides and proteins. We sought to evaluate the reproducibility and accuracy of DIA-MS metaproteomic measurements relative to DDA-MS using a mock community of known taxonomic composition. Artificial microbial communities of known composition were analyzed independently in three laboratories using DDA- and DIA-MS acquisition methods. DIA-MS yielded more protein and peptide identifications than DDA-MS in each laboratory. In addition, the protein and peptide identifications were more reproducible in all laboratories and provided an accurate quantification of proteins and taxonomic groups in the samples. We also identified some limitations of current DIA tools when applied to metaproteomic data, highlighting specific needs to improve DIA tools enabling analysis of metaproteomic datasets from complex microbiomes. Ultimately, DIA-MS represents a promising strategy for MS-based metaproteomics due to its large number of detected proteins and peptides, reproducibility, deep sequencing capabilities, and accurate quantitation. http://dlvr.it/TDYzpz
0 notes
Text
Fwd: Course: Online.Metabarcoding.2Places.Oct7-10
Begin forwarded message: > From: [email protected] > Subject: Course: Online.Metabarcoding.2Places.Oct7-10 > Date: 23 September 2024 at 05:44:48 BST > To: [email protected] > > > > > Can you please repost letting people know we onl;y have 2early bird places left for our new course on Metabarcoding and Metagenomics, if you would be > kind enough to post the followingto thecourses and workshop page of Evoldir please, Thank you, Oliver > > ONLINE COURSE – Introduction to Metabarcoding and Metagenomics Analysis > (IMAM01) > > ONLY 2 X EARLY BIRD PLACES LEFT > PR stats have added 10 early birdplaceswith 10% off reducing fees to > £432.00. The first 10 tickets are first come first serve basis when > you book via our website* > > https://ift.tt/g8bBok7 > > Instructor- Edinburgh Genomics > > 7th - 10th October 2024 > > Please feel free to share! > > COURSE OVERVIEW-Metabarcoding and metagenomics study genetic > material recovered from environmental samples. Both methods provide > a comprehensive view of microbial communities which are present in > various ecosystems. The ability to identify organisms from traces of > genetic material in environmental samples has reshaped the way we see > life on earth. Especially for microorganisms, metagenomic techniques > have granted us unprecedented insight into the microbiome of animals > and the environment more broadly > > Metabarcoding and metagenomics are both methods to study the composition > of these complex communities. Where metabarcoding focusses on looking > at a single or a combination of marker genes, metagenomics looks into > everything within a community. > > During this course we will look at the differences and similarities > between these two methods. We explain how to process the data using both > short and long reads data, we take a look at the pros and cons and some > of the pitfalls. We will guide you through the different approaches to > take when processing the data and walk you through using some of the > tools which are considered to be golden standard in the field. You will > have hands on experience processing real data. > > By the end of the course, participants should: > > Understand the basic concepts behind metabarcoding and metagenomics Work > with both short and long read data for both metabarcoding and metagenomics > Be able to use Qiime2 and NanoClust for analysis of metabarcoding Know > different methods (metaphlan, humann) for marker based taxonomic and > functional annotation of metagenomics data Create and annotated metagenome > assembled genomes (using megahit, checkm, gtdb-tk) Be able to annotated > antibiotic resistance genes in metagenomics data > > Please [email protected] any questions. > > A full list of our live courses can be found here > > > > Best wishes, > > Oliver > > Oliver Hooker PhD. > PR stats > > Oliver Hooker
0 notes
Text
Microbes of sponges have diverse associations, including true symbiosis. Sponges, being evolutionarily ancient sessile filter feeders, host diverse and abundant microbial species that play crucial roles in host metabolism. Although the microbial symbionts of sponges are widely distributed within the organism (up to 40% of their volume), the ecological relationships and interactions between bacteria and their sponge host remain largely unexplored for many species. The present study was one of the first attempts to isolate symbiotic bacteria from the sponge Raspaciona aculeata.
Materials and Methods: After isolation on marine agar medium, the isolates were characterized for different colony morphology. The 16S rDNA taxonomic analysis was carried out on bacteria isolates.
Results: Following an incubation period of two weeks at 25°C, only 13 bacterial strains were isolated with a very low rate of genetic biodiversity. All strains belonged to the Gammaproteobacteria class (Pseudomonadaceae family), except one (isolate AL-18ra) belonging to the Bacilli class (Bacillaceae family).
Conclusion: The obtained results are of great importance for advancing the understanding of symbiosis phenomena within the sponge species Raspaciona aculeata to study its bioapplication potential.
#translation #appl #research #biotechnology #biodegradation
instagram
0 notes
Quote
Metagenomes encode an enormous diversity of proteins, reflecting a multiplicity of functions and activities1,2. Exploration of this vast sequence space has been limited to a comparative analysis against reference microbial genomes and protein families derived from those genomes. Here, to examine the scale of yet untapped functional diversity beyond what is currently possible through the lens of reference genomes, we develop a computational approach to generate reference-free protein families from the sequence space in metagenomes. We analyse 26,931 metagenomes and identify 1.17 billion protein sequences longer than 35 amino acids with no similarity to any sequences from 102,491 reference genomes or the Pfam database3. Using massively parallel graph-based clustering, we group these proteins into 106,198 novel sequence clusters with more than 100 members, doubling the number of protein families obtained from the reference genomes clustered using the same approach. We annotate these families on the basis of their taxonomic, habitat, geographical and gene neighbourhood distributions and, where sufficient sequence diversity is available, predict protein three-dimensional models, revealing novel structures. Overall, our results uncover an enormously diverse functional space, highlighting the importance of further exploring the microbial functional dark matter.
Unraveling the functional dark matter through global metagenomics | Nature
0 notes
Text
Metagenomics Market Size 2022 Growth Share, Industry Dynamics, Top Trends and Regional Analysis Forecast to 2030
According to a recently released report by Future Market Insights, widespread adoption of metagenomics tools in environmental monitoring and pollution reduction projects is estimated to boost the metagenomics market growth. Recent advancements in technology are aiding in the substantial reduction of costs incurred for sequencing paving way for the development of genetic sequencing on a large scale. This has opened up new opportunities for leveraging metagenomic tools on a large scale for monitoring of environmental microbe communities. Moreover, the efficacy of metagenomic tools in tackling environmental toxicology is further contributing to its growing popularity. For instance, a recent development saw the use of metagenomics in predicting the extent and presence of contamination in the environment while another research outlined how metagenomics can be leveraged to attenuate pollutants in the environment using unculturable microbes present in the natural environment. The use of metagenomics to understand the influence biostimulation can have on microbial communities is also under progress.
Growing concerns about environmental conditions are prompting authorities around the world to invest in researches to improve the efficiency and scale of bioremediation. In addition to this, bioremediation is gradually gaining traction owing to its cost-effective and eco-friendly chemical treatment. As metagenomics is vital to the success of bioremediation, the market is estimated to grow as the alternative chemical treatment process gains popularity.
Clinical Diagnostics to Emerge as a Vital Revenue Pocket
Clinical diagnostics and bioremediation are estimated to emerge as the highest revenue generating segments in the foreseeable future. The term metagenomic diagnostics is slowly gaining in prominence as studies experiment to widen the scope of application of metagenomics in the healthcare industry. For instance, multiple studies have alluded to the development of a new diagnostic procedure termed as “shotgun metagenomics” which uses DNA sequencing to identify different pathogens and aid in the development of effective targeted treatment by predicting their resistance against different medications. Metagenomic diagnostics presents a promising prospect that could help predict and manage the treatment of a plethora of diseases. Further, metagenomics can also potentially discover hidden genetic features that could prove vital in the progression of an assortment of biotechnological applications such as the discovery of novel enzymes, bioactive molecules, and genes with significantly better or altogether new biochemical functionalities.
Metagenomics Market Growth Underpinned by its Potential Application in Antibiotics Production
Metagenomics provides a detailed analysis of the taxonomic composition of microbe communities. It provides accurate information with regards to the gene characteristics specific to a particular community. The information provided by metagenomic tools can be effectively utilized for the production of an assortment of antibiotics. Recent research in the direction was conducted by scientists at UC Berkley who concluded leveraging metagenomics for bioprospecting soil can aid in the development of a plethora of antibiotics. The study used metagenomics to identify the genes that produce antibiotics and antifungals to combat diseases in microorganisms and stated that the same chemicals can potentially aid in combating bacterial and fungal infections in humans as well.
For more information: https://www.futuremarketinsights.com/reports/metagenomics-market
Library Preparation Kits find Growing Consumer Base as Gene Sequencing Gains Centerstage
DNA and RNA sequencing practices are gradually gaining traction owing to their ability to provide a massive set of valuable data about gene sequences in a small duration of time. The data generated through gene sequencing is then analysed by clinicians, scientists, and researchers who then used the derived knowledge in various verticals such as agriculture, forensics, diagnostics, pharmaceuticals, and other applications. Library preparation kits provide the means for efficient and quick extraction of gene segments which are then used for analysis. Moreover, advancements in technology are aiding library kit manufacturers in offering improved products that reduce error and stress while improving accuracy. The FMI report opines the factors will prove vital in the growth of the segment which is estimated to contribute significantly to the metagenomics market growth.
Metagenomics Market Continues to Propel in North America, Upheld by Government Funding for Epigenetics R&D
The FMI study opines metagenomics market in North America is estimated to continue to proliferate and remain at the helm of the global metagenomics market growth. Presence of advanced infrastructure coupled with continuous government funding for research and development in the field of epigenetics are the vital factors that are estimated to drive the growth of the market in the region. Further, the presence of favorable regulations promoting research and development for improving the quality of healthcare in the countries in the region are expected to fuel the market growth.
0 notes
Text
Archaea in a warming climate become less diverse more predictable
Science News from research organizations 1 2 Date: May 5, 2023 Source: University of Oklahoma Summary: Using a long-term multifactor experimental field site researchers showed that experimental warming of a tallgrass prairie ecosystem significantly altered the community structure of soil archaea and reduced their taxonomic and phylogenetic diversity. Share: advertisement FULL STORY Led by Jizhong Zhou, Ph.D., the director of the Institute for Environmental Genomics at the University of Oklahoma, an international research team conducted a long term experiment that found that climate warming reduced the diversity of and significantly altered the community structure of soil archaea. Their findings are published in the journal Nature Climate Change. At the microbiological level, life can be described as belonging to one of three kingdoms — how species are described in relation to one another. Eukarya contains complex organisms like animals and plants and microorganisms such as fungi. The other two categories, bacteria and archaea, are comprised only of microorganisms. Archaea are prevalent in a range of environments, from some of the most hostile like volcanoes and permafrost. However, archaea are also common in the human microbiome and as an important part of soil ecology. “As temperature is a major driver of biological processes, climate warming will impact various ecological communities,” Zhou said. “Based on long-term time-series data, our previous studies revealed that experimental warming leads to the divergent succession of soil bacterial and fungal communities, accelerates microbial temporal scaling, reduces the biodiversity of soil bacteria, fungi and protists, but increases bacterial network complexity and stability. However, how climate warming affects the temporal succession of the archaeal community remains elusive. Archaea are ubiquitously present in soil and are vital to soil functions, e.g., nitrification and methanogenesis.” Using a long-term multifactor experimental field site at OU’s Kessler Atmospheric and Ecological Field Station, the researchers showed that experimental warming of a tallgrass prairie ecosystem significantly altered the community structure of soil archaea and reduced their taxonomic and phylogenetic diversity. In contrast to the researchers’ previous observations in bacteria and fungi, their finds show that climate warming leads to convergent succession of the soil archaeal community, suggesting archaeal community structures would become more predictable in a warmer world. advertisement Story Source: Materials provided by University of Oklahoma. Original written by Chelsea Julian. Note: Content may be edited for style and length. Journal Reference: Ya Zhang, Daliang Ning, Linwei Wu, Mengting Maggie Yuan, Xishu Zhou, Xue Guo, Yuanliang Hu, Siyang Jian, Zhifeng Yang, Shun Han, Jiajie Feng, Jialiang Kuang, Carolyn R. Cornell, Colin T. Bates, Yupeng Fan, Jonathan P. Michael, Yang Ouyang, Jiajing Guo, Zhipeng Gao, Zheng Shi, Naijia Xiao, Ying Fu, Aifen Zhou, Liyou Wu, Xueduan Liu, Yunfeng Yang, James M. Tiedje, Jizhong Zhou. Experimental warming leads to convergent succession of grassland archaeal community. Nature Climate Change, 2023; DOI: 10.1038/s41558-023-01664-x Cite This Page: University of Oklahoma. “Archaea in a warming climate become less diverse, more predictable.” ScienceDaily. ScienceDaily, 5 May 2023. . University of Oklahoma. (2023, May 5). Archaea in a warming climate become less diverse, more predictable. ScienceDaily. Retrieved May 5, 2023 from https://ift.tt/v9ZtBFp University of Oklahoma. “Archaea in a warming climate become less diverse, more predictable.” ScienceDaily. https://ift.tt/v9ZtBFp (accessed May 5, 2023).
0 notes
Text
What does the Name Change Entail? Differentiation of Strains for Bacterial Names

Abstract
The frequent change of bacterial names and the consequences have been discussed in brief, with recent examples of Bacillus spp, and Lactococcus spp.
Keywords: Bacterial classification, B. paralicheniformis, B. licheniformis, L. plantarum and L. pentosus
Opinion
The bacterial classification change of names “Bacillus licheniformis into B. paralicheniformis, Lactobacillus plantaruam, L. pentosus and L. paraplantarum” and implications. The change of a strain name creates many hurdles in representing, patenting, and in academic events. Even more important, patents filed under the previous name may disappear as the new name appears. The incorporation of a new name locally is easy, but this is a major concern globally. In 2019, we filed a patent on the Bioprocess developed for the purification of (B. licheniformis), bacteriocin as a result, the patent granted. During the submission of the whole genome sequence (WGS) in the NCBI portal, we were informed that this is B. paralicheniformis, but based on 16S rRNA sequence it was B. licheniformis. Identical incident observed involving L. plantarum, after submitting the genome sequence, it was informed as L. pentosus. Current microbial classifications based on 16S rRNA gene sequences, and also a few housekeeping genes [1-4] have several limitations. They begin with low phylogenetic resolution at various taxonomic ranks [5], followed by missing diversity because of primer mismatches [6], finally formation of corrupt tree topologies by drawing together various disparate groups [7]. Subsequently, we discovered the incidents that led to a change in the names of a few bacterial strains. It was a certain type of nucleotide changes to non-standard housekeeping genes such as rec A. Surprisingly, if the 99.9 % similarities found in the 16S rRNA right away it may be sequence blasted at nucleotide level and given a specific name. If not, above 99.0 % sequence similarity may be considered as another but, without considering the 16S rRNA. The dependence more on other than 16S rRNA sequence may be followed/considered/ ignored. These kinds of sudden changes in the name, causes lot of complications. The change in the name sometimes disqualifies the strain for human applications, as it has not listed in the local food safety guidelines. The name changes also created irreparable damage in commercializing the products. At the end, we should understand and accept that nothing changes the nature of the objects under classification the level of variations needs accounted for in any proposal. The classification scheme could be measured based on the number of people subscribe to it. Therefore, we may conclude that classification schemes of changing the names are rarely “right” or “wrong” but considered as simple and formal procedures to relax the complexity. This subsequently, provides a common acceptable nomenclature.
To Know More About Nutrition and Food Science International Journal
Please click on: https://juniperpublishers.com/nfsij/index.php
For more Open Access Journals in Juniper Publishers
please click on: https://juniperpublishers.com/index.php
#food biotechnology#food toxicology#Mass spectrometry in food technology#Juniper Publishers#juniper publishers open access
0 notes
Text
Abstract
Abundance and Diversity of Oyster Microbiomes from Seven Sites in the Hudson River Estuary
The eastern oyster (Crassostrea virginicia) is an important keystone and indicator species that inhabits marine ecosystems. However, the past few decades have raised concern regarding the anthropogenic impact on oyster populations. This study investigates the pathogens present within oyster microbiomes at different restoration stations in New York and New Jersey that possess varying environmental conditions such as sediment type, water temperature, and ecosystem type. Oysters were collected at the monitoring stations and stored in a freezer at -80° C until DNA extraction was conducted. Oysters were shucked to collect their tissues and either the phenol-chloroform protocol or the E.Z.N.A. Mollusc DNA Kit (Omega Bio-Tek) was utilized. The full length bacterial ribosomal RNA gene was amplified by PCR and confirmed by gel electrophoresis. Amplicons were sequenced on a Nanopore MinION according to the manufacturer’s instructions. Sequences were quality filtered and taxonomic identification was made using MIrROR (Microbial Identification using rRNA Operon Region; Seol et al. 2022). Oysters found in sandy sediment located near bays during October of 2021 had a low total reads as opposed to other areas. Using Shannon’s Diversity Index, oyster microbiomes from lagoons with muddy sediment exhibited a low species diversity despite a high read count. This may have resulted from little circulation in the water leading to minimal diversity. These findings suggest that certain environmental conditions may influence oyster microbiome health. Future research could investigate other environmental parameters such as pH, turbidity, and oxygen concentration of water.
Keywords: eastern oyster, environmental conditions, anthropogenic impact, microbiome, Shannon’s Diversity Index
0 notes
Text
Characterization of a marine bacteria through a novel metabologenomics approach
Exploiting microbial natural products is a key pursuit of the bioactive compound discovery field. Recent advances in modern analytical techniques have increased the volume of microbial genomes and their encoded biosynthetic products measured by mass spectrometry-based metabolomics. However, connecting multi-omics data to uncover metabolic processes of interest is still challenging. This results in a large portion of genes and metabolites remaining unannotated. Further exacerbating the annotation challenge, databases and tools for annotation and omics integration are scattered, requiring complex computations to annotate and integrate omics datasets. Here we performed a two-way integrative analysis combining genomics and metabolomics data to describe a new approach to characterize the marine bacterial isolate BRA006 and to explore its biosynthetic gene cluster (BGC) content as well as the bioactive compounds detected by metabolomics. We described BRA006 genomic content and structure by comparing Illumina and Oxford Nanopore MinION sequencing approaches. Digital DNA:DNA hybridization (dDDH) taxonomically assigned BRA006 as a potential new species of the Micromonospora genus. Starting from LC-ESI(+)-HRMS/MS data, and mapping the annotated enzymes and metabolites belonging to the same pathways, our integrative analysis allowed us to correlate the compound Brevianamide F to a new BGC, previously assigned to other function. http://dlvr.it/TBpG2S
0 notes
Text
Fwd: Course: Online.ShotgunMetagenomics.Dec10-13
Begin forwarded message: > From: [email protected] > Subject: Course: Online.ShotgunMetagenomics.Dec10-13 > Date: 29 August 2024 at 05:44:19 BST > To: [email protected] > > > > Dear all, > > We are excited to announce our upcoming online course, "Shotgun > Metagenomics Processing and Analysis," taking place from December > 10-13. This course is designed to provide a comprehensive overview of > advanced methodologies for studying microbial communities using shotgun > metagenomics data. > > Course website: ( > https://ift.tt/S3b8LQV ) > > Course Overview: Participants will engage in both lectures and hands-on > lab sessions to tackle key challenges in metagenomics, including: Data > preprocessing Taxonomic profiling Strain-level characterization Microbial > genome reconstruction Functional potential characterization Integrative > statistical data analysis By the end of the course, attendees will gain > familiarity with cutting-edge bioinformatics tools and visualization > techniques essential for metagenomics research. > > Target Audience: This course is ideal for researchers and students > looking to enhance their skills in analysing high-throughput microbiome > data. A basic knowledge of the command line is recommended; we suggest > completing an online tutorial if you're new to this. > > Learning Outcomes: Understanding the goals and approaches in studying > microbial communities Proficiency in taxonomic, gene-related, and > strain-level characterization using reproducible workflows Expertise in > statistical analyses and visualization tools for microbiome studies > > For the full list of our courses and workshops, please visit: ( > https://ift.tt/S3b8LQV ) > > Best regards, > > Carlo > > > Carlo Pecoraro, Ph.D > Physalia-courses DIRECTOR > [email protected] > mobile: +49 17645230846 > > > "[email protected]"
0 notes
Text
#cancers, Vol. 16, Pages 1923: Exploring Gut Microbiome Composition and Circulating Microbial DNA Fragments in Patients with Stage II/III Colorectal #cancer: A Comprehensive Analysis
Background: Colorectal #cancer (CRC) significantly contributes to #cancer-related mortality, necessitating the exploration of prognostic factors beyond TNM staging. This study investigates the composition of the gut microbiome and microbial DNA fragments in stage II/III CRC. Methods: A cohort of 142 patients with stage II/III CRC and 91 healthy controls underwent comprehensive microbiome analysis. Fecal samples were collected for 16S #rRNA sequencing, and blood samples were tested for the presence of microbial DNA fragments. De novo clustering analysis categorized individuals based on their microbial profiles. Alpha and beta diversity metrics were calculated, and taxonomic profiling was conducted. Results: Patients with CRC exhibited distinct microbial composition compared to controls. Beta diversity analysis confirmed CRC-specific microbial profiles. Taxonomic profiling revealed unique taxonomies in the patient cohort. De novo clustering separated individuals into distinct groups, with specific microbial DNA fragment detection associated with certain patient clusters. Conclusions: The gut microbiota can differentiate patients with CRC from healthy individuals. Detecting microbial DNA fragments in the bloodstream may be linked to CRC prognosis. These findings suggest that the gut microbiome could serve as a prognostic factor in stage II/III CRC. Identifying specific microbial markers associated with CRC prognosis has potential clinical implications, including personalized treatment strategies and reduced healthcare costs. Further research is needed to validate these findings and uncover underlying mechanisms. https://www.mdpi.com/2072-6694/16/10/1923?utm_source=dlvr.it&utm_medium=tumblr
0 notes
Text
Researchers Examine Changes to the Microbiota Composition and Metabolic Profiles of Patients with Recurrent Clostridium difficile Infection (rCDI) Following Treatment with Faecal Microbiota Transplant (FMT)
Researchers Examine Changes to the Microbiota Composition and Metabolic Profiles of Patients with Recurrent Clostridium difficile Infection (rCDI) Following Treatment with Faecal Microbiota Transplant (FMT)
Microbial taxonomic and metabolic alterations during faecal microbiota transplantation to treat Clostridium difficile infection
,
Gwénaëlle Le Gall1,

Email the author Gwénaëlle Le Gall
,
Marianne Defernez
Email the author Marianne Defernez
,
Ian L.P. Beales
Email the author Ian L.P. Beales
,
Ngozi Franslem-Elumogo
Email the author Ngozi Franslem-Elumogo
,
Arjan Narbad
Email the author Arjan Narbad
View On WordPress
#C.difficile infection research#CDI research#Clostridium difficile Research#faecal microbiota transplant (FMT).#Microbial taxonomic
0 notes
Text
Do viruses “hijack” the body? “Viruses are more like cone snails than hijackers.” Cone shell venom, weaponized hormones, and predatory insulin overdose. Gila monsters and diabetes treatment. Mimicry. Totally unlike beings contain patches of intimate and detailed sameness. Bodies hold imprints of the other entities’ influence, like a shadow. Where does one creature end and another creature begin?
---
In the twentieth century, the word “hijacking” came to typify the explanation of what viruses do to the cells they infect. How commonplace it is to now say: the virus hijacks the host cell’s machinery in order to replicate itself. [...] However, viruses are not like spaceships, and cells are not just like twentieth-century semitrailer trucks, armored vehicles, or passenger jets whose resources can be plundered and whose operators can be coerced into unwanted journeys. [...] It appears to transparently explain things. But is this a good description of a virus and the creatures it is capable of infecting? [...]
Anthropomorphism and personification of microbial entities in the explanation of virology is a understandable tendency; in this way, viral action is domesticated to the human scale.
But maybe it’s time to practice some resolutely non-domesticating conid-amorphism, some conus-centrism, and see what happens if we forge a new avenue for thinking of viruses in terms of venomous cone snails.
---
Conidae is the taxonomic name for the family of gastropods known more colloquially as cone snails, or predatory sea snails. “One of the most successful lineages of marine animals,” the hundreds of species of Conidae are characterized by their use of complex venoms to capture prey. Some of these snails prey on worms, some on other snails, and some on fish. In 2015, researchers reported that Conus geographus uses an insulin overdose to disorient and disable its fish prey, releasing the toxin into the water.
Insulin appears to be a component of the nirvana cabal, a toxin combination in these venoms that is released into the water to disorient schools of small fish, making them easier to engulf with the snail’s distended false mouth, which functions as a net. If an entire school of fish simultaneously experiences hypoglycemic shock, this should directly facilitate capture by the predatory snail.
The released insulin does not affect the snail itself, because its venom insulin mimics fish insulin, not its own molluscan variety. Venomous snails that hunt worms in this fashion make a different insulin mimic, specific to worms.
---
This may seem like a weird aberration on the part of an obscure life-form, yet cone snails are only one kind of creature that roams the evolutionary space in which one being’s molecules evolve to participate in the physiology of another. There are enough examples of organisms employing these tactics that they are grouped together in scientific reviews of “weaponized hormones.” [...]
For example, the first diabetes therapy drug that works by mimicking the glucagon-like peptide hormone (GLP-1) was discovered by analyzing the venom of the Gila monster. GLP-1 is a hormone released by the gut after eating that stimulates insulin production and slows movement of food through the intestinal tract. The making of a drug that acts like GLP-1 was not a process of exactly copying human GLP-1. For that you wouldn’t need a Gila monster and thousands of years of coevolution between lizards and their prey.
Rather, scientists learned alternative molecular strategies for binding to the hormone’s receptor from the Gila monster venom component.
Perhaps viruses are equally “interested” in glucose control in their own noncognitive way, in that their replication and continued existence is vested in their hosts’ metabolisms. As noted above, they do not possess the means for making the proteins encoded by the genes in their genomes, but rather depend on the cells they infect to do that translation and transcription. [...]
They don’t have their own bodies; they only have their hosts’ bodies.
And bit by bit, through the endless nonillion-fold exploration of evolutionary space -- the space that makes the difference between persisting long enough to be replicated versus falling apart into the wash of biological decay -- it has turned out to be good practice to mimic your host’s cell cycle and metabolic hormones. Different viruses, different hosts, same strategy. [...]
---
If viruses are effective because they mimic human hormones or hormone receptors, this will drive the evolution and duplication of hormones over time because it is advantageous for them to change enough to not be good viral targets. Thus, it has been suggested that some features of human hormonal genes in the placenta arise from their function as viral “decoys” in this tit-for-tat of copying and changing.
It is for this reason that viruses could be thought of in terms of venomous cone snails rather than hijackers.
Not all these mimicry techniques target metabolic processes; many viruses famously encode other proteins that allow them to evade immune detection by attempting to look like parts of the immune system rather than its targets. But there is a theme here. The virus takes on some aspects of the shape of the body that ensures its continuity. This gives rise to totally unlike beings containing patches of intimate and detailed sameness, molecules that bear the same precise contours to fit into a particular hormone receptor, yet perhaps bear no common genetic heritage. Viruses, like cone snails, evolve to be more like what sustains them. It is an uncomfortable form of relatedness, this predatory metabolic convergence, but it cannot be denied that it generates amazing patterns of likeness across biological kingdoms without everything having to be descended from the same line of direct genetic inheritance.
Where does hormone end and hormone-like begin?
If the mimic converges on the original, and places the original under evolutionary pressure to diverge, what is left is a seesawing mirrored relationship of competitive difference and similarity, not an original and a mimic. Even if something has evolved to get away from its mimic, it holds the imprint of that entity’s influence in its difference, like a shadow.
In practical terms, looking into cone snails and viral genomes suggests new ways of making drugs, which are human-made mimics that seek to manipulate physiology by augmenting or suppressing the action of the molecule that has been mimicked.
In philosophical terms on the other hand, that cone snail or amphibian venom gets enrolled in the diabetes epidemic by becoming a blood sugar–controlling drug indicates that anthropomorphic concepts such as hijacking may possibly be the least illuminating explanatory tactic for understanding what viruses do, and are, in relation to their bodies. (Yes, their bodies. That means us).
---
Text by: Hannah Landecker. “Viruses Are More Like Cone Snails Than Hijackers.” e-flux. October 2022. [Bolded emphasis and italicized first paragraph added by me.]
70 notes
·
View notes
Text
Lupine publishers|Variability of Chromophytic Phytoplankton in the Pacific Ocean and Indian Ocean: A Review
Variability of Chromophytic Phytoplankton in the Pacific Ocean and Indian Ocean: A Review

Abstract Generally, phytoplankton exhibits high species diversity and wide range of variation which makes them most successful primary producer in contemporary global oceanographic regimes. When compared to its counterpart of terrestrial ecosystem, phytoplankton species diversity is twelve-fold lower, however, taxonomic divisions are eight times higher than terrestrial plants. Therefore, it is important to investigate the detailed composition of phytoplankton and study their spatiotemporal distribution in the marine environment. In the present review, we have assessed the detailed community composition of cormophytic phytoplankton with their variability in the world oceans
Introduction In aquatic ecosystem, primary production mostly depends on the photosynthetic process carried out by autotrophic organisms which include phytoplankton, phytobenthos and macroalgae. Although phytoplankton are responsible for only 1-2% of the total global biomass, they produce 30-60% of the global annual fixation of carbon on Earth. This ultimately leads to providing the necessary energy for consumers and ultimately, to human beings. In marine ecosystem primary productivity is mainly focused in euphotic zone and marine phytoplankton are the key players which carry out carbon fixation via photosynthesis. Generally, phytoplankton community structure is characterized by suits of approaches including light microscopy, flow cytometry and pigment analysis. Recently, new technologies such as molecular methods (Next generation sequencing and 18s rRNA) have revolutionized the characterization of phytoplankton. However, due their evolutionary conserved nature and multiple copies, 18s rRNA may not provide high resolution and sometimes fail to identify phytoplankton at species or at genus level. Whereas, functional genes are based on selected functional aspect of organisms and achieve greater resolution of community structure. Moreover, these functional genes provide insights of element metabolism (e.g. carbon and nitrogen), which are potentially linked to biogeochemical cycling in oceans. Recently, functional gene markers including rbcL (encoding RuBisCO enzyme), increasingly being used in deciphering community composition of chromophytic phytoplankton. Generally, RuBisCO enzyme involved in Calvin-Benson cycle which assimilate carbon dioxide to organic carbon. RuBisCO contains four different forms (Form I, II, III and IV). Form I contain 8 large and 8 small (L8S8) subunits. It is mainly found in all plants and few bacteria. Whereas form II (L2) is found in Dinoflagellates and few bacteria. Form III has been recovered from some archaea, while form IV which is also known as RuBisCO-Like Protein (RLP) is identified from variety of microbial group. Among all these forms, large subunit is widely distributed in all the four forms. It contains more conserved sequences making it a suitable gene marker for phylogenetic analysis. Specifically, Form I RuBisCO gene is divded into green and red lineages whereas, non-green phytoplankton contain form ID RuBisCo and termed as chromophytic phytoplankton. Numerous previous studies based on large subunit have recognized its importance in decrypting community structure of chromophytic phytoplankton and their vast distribution in different ecological settings of world oceans. Pacific Ocean Pacific Ocean is the largest ocean and numerous studies have been carried on chromophytic phytoplankton. Generally, studies were carried out at east Pacific Ocean, north Pacific Ocean and west Pacific Ocean. In the region of west Florida continental shelf, the study carried out by Pichard et al. [3] on Form I rbcL gene showed dominance of Prochlorococcus and Synechococcus in euphotic zone. However, in the in deep euphotic zone, they observed manganese- oxidizing bacterium, ultimately suggesting that chemoautotrophs might have contributed to the diversity of carbon fixing organisms in the marine euphotic zone. Another study carried out at costal part of west Pacific Ocean i.e., Monterey Bay costal upwelling zone by Bhadury et al. [2], showed dominance of bloom forming Diatoms including Pseudo-nitzschia sp., and Thalassiosira sp. Moreover, they have also observed the dominance of Haptophycea groups such as Emiliania huxleyi. However, this study concluded that obvious environmental parameters such as temperature and nitrate did not strongly correlate with species abundance. Contrast to these results of upwelling zone, the study carried out by our group Pujari et al. [3] Accepted
and in printing) showed strong correlation to the most of environmental parameters present in the mindano upwelling zone located at West Pacific Ocean. West Pacific Ocean is composed of western boundary currents which play the key role in maintaining world climate. These currents are the most dynamic and intensive mass water transportation system in the world. Our study showed that warmer, low saline and nutrient limited conditions regulated the Cyanophyceae (mainly Prochlorococcus) group in surface and subsurface depths. Further, we also observed dominance of Haptophyceae group species such as Chrysochromulina dominated the deeper depths which were correlated with high salinity and fairly high concentration of dissolved nutrients. Moreover, one of the key species of upwelling zone was dominated by Pelagomonas of Pelagophyceae group. It was influenced by availability of upwelled nutrients, and adapted to light limited conditions. Similarly, one of the studies based on monthly observation for two years carried out in Station ALOHA and North Subtropical Gyre (NPSG) of Pacific Ocean by Li et al. [5] showed that despite of prevailing oligotrophic conditions, diatoms, prymnesiophytes and dinoflagellates were dominant with varying concentration. qPCR analysis showed that these groups gene abundance often varied more than order of magnitude between successive months. Specifically, Diatoms rbcL gene abundance was more apparent at upper and lower regions of the euphotic zone. Whereas Prymnesiophytes and Pelagophytes were significantly high in number at lower euphotic zone than in the upper euphotic zone. One of the iron enrichment experimental studies based on quantification of rbcL transcripts using qRT-PCR by Endo et. al. [6] in Bering Sea of Northern Pacific Ocean showed that, regardless of Fe availability, the transcript abundance of rbcL gene decreased in the high CO2 treatments (600 and 1000 ppm). This ultimately showed that projected future increase in seawater pCO2 could reduce the RuBisCO transcription in Diatoms, resulting in decreased primary productivity and there could be possible shift in the food web structure of the Bering Sea. Indian Ocean Indian ocean is the third largest ocean in the world. And covers almost 20% of the water on Earth’s surface. Within Indian ocean, there are few marginal seas, including Bay of Bengal, Arabian Sea, Laccadive Sea, Somalia Sea and Andaman Sea. Although Indian Ocean is one of the largest seas, it is significantly ignored for the distribution and ecology of chromophytic phytoplankton. Only few studies are carried out in the Indian Ocean region. One of such study was carried out by our group, Pujari et al. [4] in the Bay of Bengal. This study serves as the first report of chromophytic phytoplankton from that region. Study was carried out with the combination of techniques (molecular and traditional taxonomy using stereo microscope) to decipher the community structure of chromophytic phytoplankton. Our study observed variation in spatial distribution of chromophytic phytoplankton using rbcL genes and morphologically identification which was likely impacted by coastal freshwater input, winter monsoons, and upwelling. Moreover, as this study report first molecular related characterization of chromophytic phytoplankton, numerous new lineages were reported. One of such genus was Bolidomonas which ubiquitously distributed but only constitute minor component of phytoplankton community. Bolidomonas is a unicellular alga and with the isolated strains by comparing nuclear, plastidal and mitochondrial gene markers, Ichinomiya et al. [7] combined Bolidomonas and Triparma and included them into Parmales. Further, phylogenetic analysis showed that Parmales (Bolidophyceae) are closely related to Diatoms Unlikely open oceans
or coastal regions, one of the studies was carried out by Samanta and Bhadury, (2014) in the Sundarbans mangrove ecosystem of the Bay of Bengal for decrypting spatiotemporal community composition of chromophytic phytoplankton. The Sundarbans which is the part of the largest deltaic mangrove ecosystem located at the apex of the Bay of Bengal. It encompasses over 102 islands with a network of countless rivers, rivulets and creeks. Bay of Bengal is vast deltaic region which covers almost area of 10000 km2, and generally it is influenced by the coastal waters from the Bay of Bengal. As observed in other mangrove ecosystem of South Asia and South East Asia, diatoms outnumber other chromophytic phytoplankton assemblages. Similarly, Samanta et al. [8] observed by far high clone libraries of Diatoms (Bacillariophyceae). Moreover, for the first time from Sundarbans mangrove ecosystem, this study detected other chromophytic phytoplankton community, including Cryptophyceae, Haptophyceae,Pelagophyceae, Eustigmatophyceae and Raphidophyceae. To summarize, numerous studies were undertaken for deciphering community composition of chromophytic phytoplankton. Moreover, as we are progressing towards new technologies to assess the diversity of these phytoplankton, new species are being discovered. Many recent studies are being able to identify the new species which were not reported in the particular niche.
For more information about Modern Approaches in Oceanography and Petrochemical Sciences archive page click on below link
https://lupinepublishers.com/ocean-journal/archive.php
For more information about lupine publishers page click on below link
https://lupinepublishers.com/index.php
#lupine publishers group#Modern Approaches in Oceanography and Petrochemical Sciences#phylogenetic analysis
2 notes
·
View notes