#mdd to new mdr classification
Explore tagged Tumblr posts
Visit Tumblr Blog
Explore Tumblr blogs with no restrictions, modern design and the best experience.
Last Seen Tumblr Blogs





Fun Fact
Tumblr.com is the 103rd most visited website in the world.
Text
What You Need to Know About Medical Device Regulation in Europe
Introduction about Medical Device Regulation in Europe
Introducing the Medical Device Regulation in Europe addresses several challenges. It aims to enhance medical device regulation in Europe to ensure a higher level of safety and effectiveness. The need for the MDR arises from various factors, including shortcomings identified in the previous regulatory framework (Medical Device Directives) and the evolving landscape of medical technologies.
Rapid technological advancements in the field of medical devices have led to the development of increasingly complex and innovative products. The MDR adapts the regulatory framework to account for these technological advancements and to ensure the safety and performance of new and emerging devices.
The primary objective of the MDR is to prioritize patient safety. By introducing more stringent requirements for conformity assessment, clinical evidence, and post-market surveillance, the regulation aims to reduce the risk of harm to patients and users of medical devices. The MDR promotes transparency by introducing measures such as the European Database on Medical Devices (EUDAMED).
This database allows better traceability of medical devices in the market and facilitates communication between regulatory authorities, manufacturers, and other stakeholders. The MDR places a greater emphasis on post-market surveillance activities, ensuring that medical devices are continually monitored once they are on the market.
This enables timely identification and response to safety issues and improves overall device performance.
The MDR aims to harmonize the regulatory requirements for medical devices across EU member states. This harmonization facilitates a more consistent and predictable regulatory environment for manufacturers, streamlining the process of bringing products to market.
The MDR introduces a more refined classification system for medical devices, taking into account the potential risks associated with devices. This allows for a more accurate categorization of devices based on their characteristics and intended use. The MDR clarifies the roles and responsibilities of economic operators, including manufacturers, authorized representatives, importers, and distributors.
This enhances accountability throughout the supply chain, ensuring that each party plays a defined role in ensuring device compliance and safety.
The MDR aligns with global best practices and standards, contributing to international regulatory convergence. This alignment is essential for manufacturers who intend to market their devices globally. The MDR addresses identified gaps and weaknesses in the previous regulatory framework, the Medical Device Directives (MDD).
These gaps included challenges related to the classification of devices, insufficient requirements for clinical evidence, and variations in the interpretation and application of the directives among member states.
Medical Device Compliance
Medical device compliance refers to the adherence of medical devices to regulatory standards and requirements set by relevant authorities. Ensuring compliance is crucial to guarantee the safety, efficacy, and quality of medical devices, protecting both patients and healthcare providers.
The key aspects of medical device compliance are:
Regulatory Authorities
Different countries have regulatory bodies responsible for overseeing medical devices. For example, the U.S. FDA in the US, the European Medicines Agency (EMA) in the EU, and the Pharmaceuticals and Medical Devices Agency (PMDA) in Japan. Manufacturers must comply with the regulations specific to the regions where they intend to market their devices.
Classification of Devices
Medical devices are often categorized based on their risk level. The risk classifications include Class I, IIa, IIb, and III in the European Union under the MDR.
Quality Management Systems (QMS)
Compliance often involves the implementation of a QMS, such as ISO 13485. This ensures that manufacturers establish and maintain processes to consistently produce safe and effective medical devices.
Pre-market Approval (PMA) or Conformité Européenne (CE) Marking
Before marketing a medical device, manufacturers may need to obtain regulatory approval or clearance. In the U.S., this may involve the submission of a PMA application, while in the EU, devices need to be CE marked according to applicable regulations.
Post-market Surveillance
Manufacturers are required to monitor and report adverse events and product issues after a device is on the market. This involves maintaining a system for post-market surveillance and reporting to regulatory authorities.
Labelling and Instructions for Use
Compliance includes providing accurate and clear labelling for medical devices. Instructions for use must be easily understandable, and information about potential risks and proper usage should be prominently displayed.
Risk Management
Manufacturers must conduct risk assessments for their devices to identify and mitigate potential risks throughout the product lifecycle.
Clinical Data and Performance
Submission of relevant clinical data is often required for regulatory approval. This data supports claims regarding the safety and performance of the medical device.
Adherence to Standards
Compliance with relevant industry standards, such as those developed by the International Electrotechnical Commission (IEC) or ASTM International, is often necessary to demonstrate that a device meets specific criteria.
Audits and Inspections
Regulatory authorities may conduct audits and inspections to ensure that manufacturers are complying with all applicable regulations.
EU Medical Device Directive
The Medical Devices Directive (93/42/EEC) was one of the main directives governing the marketing and distribution of medical devices within the European Union. It provided the regulatory framework for the safety and performance of medical devices and outlined essential requirements that devices needed to meet.
The MDR (2017/745) came into effect in May 2021, replacing the Medical Devices Directive. It introduced more stringent requirements for the approval and surveillance of medical devices in the European Union.
The MDR emphasizes increased transparency, traceability, and the involvement of notified bodies in the conformity assessment process.
The MDR came into force on May 26, 2017, but the date of application was postponed several times. As of my last update, it was set to be fully applicable from May 26, 2021.
Key Changes and Features:
Stricter Scrutiny: The MDR introduces more rigorous pre-market assessment procedures, including enhanced scrutiny of high-risk devices.
Unique Device Identification (UDI): It mandates the use of a UDI system for better traceability of devices throughout their lifecycle.
EUDAMED Database: The establishment of the European Database on Medical Devices (EUDAMED) for the registration and dissemination of information about medical devices.
Post-Market Surveillance (PMS) and Post-Market Clinical Follow-up (PMCF): Strengthened requirements for post-market surveillance and clinical follow-up to monitor and report on device performance.
According to the MDR 2017/745, a ‘medical device’ means any instrument, apparatus, appliance, software, implant, reagent, material or other article intended by the manufacturer to be used, alone or in combination, for human beings for one or more of the following specific medical purposes:
diagnosis, prevention, monitoring, prediction, prognosis, treatment or alleviation of disease,
diagnosis, monitoring, treatment, alleviation of, or compensation for, an injury or disability,
investigation, replacement or modification of the anatomy or of a physiological or pathological process or state,
providing information by means of in vitro examination of specimens derived from the human body, including organ, blood and tissue donations.
Medical Device Certification in Europe
The CE marking is a certification mark that indicates conformity with health, safety, and environmental protection standards for products sold within the European Economic Area (EEA). The CE marking is mandatory for various product categories, including medical devices.
For medical devices, obtaining the CE marking signifies compliance with the applicable European regulations, such as the Medical Device Regulation (MDR) or the In Vitro Diagnostic Regulation (IVDR).
The CE marking demonstrates that a product meets the essential requirements outlined in European Union (EU) directives or regulations, ensuring a high level of safety and performance. Medical devices that are intended to be placed on the market within the EEA must bear the CE marking.
This includes a wide range of products, from simple devices to complex technologies. The CE marking allows a medical device to be legally placed on the market and sold within the EEA. Various economic operators in the supply chain, including manufacturers, importers, distributors, and authorized representatives, have specific responsibilities related to the CE marking process.
As part of the new regulatory framework, information about medical devices, including their CE certificates, will be registered in the European Database on Medical Devices (EUDAMED).
Manufacturers must continuously ensure that their devices remain in compliance with regulatory requirements. This includes monitoring post-market surveillance data and promptly addressing any safety concerns.
Manufacturers of medical devices must follow a series of steps to affix the CE marking:
Conformity Assessment: The manufacturer assesses the device’s conformity with applicable EU regulations. This assessment can involve self-certification or the involvement of a Notified Body, depending on the device’s risk classification.
Technical Documentation: The manufacturer prepares comprehensive technical documentation that demonstrates compliance with essential requirements, including design, manufacturing, and performance data.
Quality Management System (QMS): Manufacturers must implement and maintain a QMS in accordance with relevant standards, such as ISO 13485.
Declaration of Conformity: Once the conformity assessment is successful, the manufacturer issues a Declaration of Conformity, declaring that the device meets the requirements of applicable EU regulations.
Affixing the CE Mark: The CE marking is affixed to the product, packaging, or accompanying documentation, indicating that the device complies with EU regulations.
Europe healthcare device standards
European healthcare device standards are a set of technical specifications and criteria that provide a common language and framework for the design, manufacturing, and performance of healthcare devices within the European Union (EU).
These standards are developed by various standardization organizations, and compliance with these standards helps manufacturers meet regulatory requirements and ensure the safety and efficacy of their devices. Some of these standards are:
ISO Standards: cover various aspects, including quality management, risk management, and specific requirements for different types of medical devices.
Electromagnetic Compatibility (EMC) Standards: Healthcare devices, especially electronic and electrical equipment, must comply to ensure that they do not interfere with other devices and are not susceptible to interference. Common standards include EN 60601-1-2 for medical electrical equipment.
Biocompatibility Standards: ensure that materials used in devices do not cause adverse reactions when in contact with the human body. EN ISO 10993 is a series of standards addressing biocompatibility.
Quality Management System (QMS) Standards: Compliance with quality management system standards is essential for medical device manufacturers. ISO 13485 is an international standard, and compliance with it is often required for the CE marking of medical devices in Europe.
Risk Management Standards: Risk management is a critical aspect of medical device design and manufacturing. EN ISO 14971 is the standard that outlines the principles for risk management.
Sterilization Standards: standards like EN ISO 11135 for ethylene oxide sterilization and EN ISO 17665 for moist heat sterilization provide guidelines for ensuring the effectiveness of the sterilization process.
Software Standards: As software plays an increasingly important role in healthcare devices, standards like IEC 62304 provide guidance on the software life cycle processes.
Usability and Human Factors Standards: Standards in this category address the design and usability of healthcare devices to ensure that they are user-friendly and safe. EN ISO 14971 and IEC 62366 are relevant standards.
Labelling Standards: Standards related to labelling provide guidance on the information that should be included on medical device labels. EN 980 and EN ISO 15223-1 are examples.
It’s important for manufacturers and stakeholders in the healthcare industry to stay updated on the latest standards and ensure compliance with relevant regulations, as these standards play a crucial role in demonstrating conformity to essential requirements for medical devices in the European market.
Additionally, the specific standards applicable to a particular device depend on its type, classification, and intended use.
Conclusion
In conclusion, the unveiling of the Medical Device Regulation in Europe represents a significant milestone in the evolution of regulatory standards for Medical Device Regulation in Europe. The comprehensive changes introduced by the MDR are driven by a commitment to advancing patient safety, fostering innovation, and adapting to the rapidly evolving landscape of healthcare technologies.
The MDR addresses critical shortcomings identified in the previous regulatory framework, offering a more robust and transparent system for the approval and surveillance of medical devices.
With a heightened focus on risk management, clinical evidence, and post-market surveillance, the MDR aims to ensure that medical devices entering the European market meet the highest standards of safety and efficacy.
Key elements of the MDR include an enhanced classification system, stricter conformity assessment procedures, and the implementation of a Unique Device Identification (UDI) system. These features contribute to a more sophisticated and nuanced approach to regulatory compliance, tailored to the diverse range of medical devices and their associated risks.
The regulation’s emphasis on transparency is evident in the establishment of the EUDAMED, providing stakeholders with a centralized platform for information sharing and increased traceability. This marks a crucial step towards creating a harmonized regulatory environment across EU member states.
As manufacturers navigate the complexities of compliance, understanding the requirements of the MDR becomes paramount. The alignment of the MDR with global standards fosters a seamless transition for manufacturers looking to market their devices internationally, reinforcing the EU’s commitment to global regulatory convergence.
In essence, the EU MDR sets a new benchmark for the medical device industry, demanding higher standards of quality, safety, and accountability. While the transition may present challenges for manufacturers, the long-term benefits lie in a safer and more innovative landscape that ultimately benefits patients and healthcare systems across Europe.
Staying informed about the intricacies of the MDR and collaborating with regulatory experts will be crucial for industry players aiming to navigate this transformative regulatory landscape successfully.
Originally Published at: https://omcmedical.com/things-about-medical-device-regulation-in-europe/
0 notes
Text
Transitioning from MDD to MDR: Navigating the Evolution of Medical Device Regulation
In the dynamic landscape of healthcare, the regulatory environment governing medical devices is undergoing a transformative shift. The transition from the Medical Device Directive (MDD) to the Medical Device Regulation (MDR) marks a significant milestone, bringing about changes in requirements, compliance, and the overall regulatory framework. This blog aims to provide insights into the key aspects of transitioning from MDD to MDR, guiding manufacturers through this pivotal phase.
Understanding the Transition:
The Medical Device Regulation (MDR), adopted in 2017 and fully applicable since May 26, 2021, replaces the previous Medical Device Directive (MDD) and the Active Implantable Medical Devices Directive (AIMDD). This transition is designed to enhance the safety and performance of medical devices while addressing technological advancements and aligning with global regulatory trends.
Key Changes and Impacts:
1. Expanded Scope:
- MDR expands the scope of regulated medical devices to include certain products that were previously not covered under MDD.
- Manufacturers need to reassess their product classifications and ensure compliance with the new criteria.
2. Stricter Clinical Evidence Requirements:
- MDR emphasizes the importance of robust clinical evidence to support the safety and performance of medical devices.
- Manufacturers must conduct thorough clinical evaluations and update their documentation accordingly.
3. Unique Device Identification (UDI):
- Introduction of a UDI system is a significant change, requiring each device to bear a unique identifier for traceability throughout its lifecycle.
- Manufacturers need to implement UDI labeling and data management systems.
4. Economic Operators and Supply Chain Responsibilities:
- MDR introduces new roles, including Importers and Distributors, each with defined responsibilities in ensuring compliance and reporting adverse events.
- Manufacturers must establish clear agreements and communication channels within the supply chain.
5. Post-Market Surveillance (PMS) and Vigilance:
- Enhanced post-market surveillance requirements necessitate proactive monitoring of device performance in real-world settings.
- Vigilance reporting obligations are expanded, requiring timely reporting of incidents to competent authorities.
6. Conformity Assessment Procedures:
- MDR introduces more rigorous conformity assessment procedures, particularly for high-risk devices.
- Manufacturers should engage with Notified Bodies early in the development process to ensure a smooth transition.
Challenges in the Transition:
1. Resource Intensity:
- The transition process demands significant resources, including time, expertise, and financial investments.
- Manufacturers must allocate resources efficiently to meet the stringent MDR requirements.
2. Limited Notified Body Capacity:
- The limited capacity of Notified Bodies for MDR certification has created challenges in scheduling assessments.
- Manufacturers need to plan well in advance and engage with Notified Bodies promptly.
3. Data Collection and Management:
- Meeting the heightened clinical evidence requirements may pose challenges in terms of data collection and management.
- A comprehensive strategy for clinical evaluations and post-market surveillance is essential.
The transition from MDD to MDR represents a paradigm shift in the regulatory landscape for medical devices. Manufacturers must proactively adapt to the changes, invest in compliance measures, and collaborate closely with regulatory authorities and Notified Bodies. By embracing these challenges as opportunities for improvement, the industry can ensure the continued delivery of safe and effective medical devices, meeting the evolving needs of healthcare and enhancing patient safety across the European market.
0 notes
Text
MDD to New MDR Classification of Medical Devices
The prerequisites for MDR classification for medical devices are basically equivalent to those in the present Medical Devices Directive (MDD). The EU MDR is shaking up the medical device industry and the order rules have not been left immaculate. The MDR decides the congruity assessment course for the device. While the order principally worries of the maker, if the device falls into Classes IIa, IIb, or III it has suggestions for the Notified body.
Before medical devices manufacturers can lawfully CE stamp their products in Europe, they should consent to the fitting medical devices order or guideline set out by the EU Commission. It is crucially critical to know the right medical device classification for your product before CE marking your devices. A classification impacts the administrative prerequisites for your devices, just as the endorsement course and its related expenses.
The number of rules in the MDR classifications has expanded from 18 to 22 and extra changes have been made to existing guidelines, which means numerous devices have new classifications. One of the important changes with the EU MDR is that medical device manufacturers will feel as they progress to conform to the new guideline is the adjustment in prerequisites for devices classifications. As medical devices classification change so do the prerequisites for manufacturers. The MDR will apply to specific products not directed through the MDD, such devices without an expected medical reason like non – corrective contact lenses.2 The MDR will likewise uncommonly manage devices joining nonmaterial’s and devices fabricated with non – feasible human tissue, which are at present absolved from the MDD.
EU MDR classifications for medical devices
One of the first considerations for medical devices manufacturers who are seeking to place their products in the European markets is to determine which the appropriate classifications for their devices are.
The MDR classification of the device will impact on how and when you will engage with your Notified Body. As the market transitions from Medical Devices Directives (MDD) and the Active Implantable Medical Devices to the Medical Devices Regulation, the device manufacturers must note the changes in the requirements for device classification. For example, devices that had previously been included in the Active Implantable Medical Device Directives (MDD) are now covered in the Medical Device Regulation (MDR).
Medical Device Classification as per MDR
As per MDR Article, 51 Medical Devices are divided into class I, IIa, IIb and class III, taking into account the intended purpose of the devices and their inherent risks. Medical Device classifications are mainly based on the following factors:
Does the device have a standalone action?
How long the device is in continuous use in the human body?
Is it an invasive device or surgically invasive Medical Device?
Is it an implantable or active medical device
Does the Medical Device serve the purpose by the use of a certain drug?
EU MDR Medical Device Classification Rules The Medical Device Classification EU rules, which are based on the vulnerability of the human body, should take into account the potential risks associated with the technical design and manufacture of the devices. Read more - MDR classification Visit Us - Operon StrategistContact details – Phone no - 93702 83428 Mail id – [email protected]
#mdd to mdr#medical device regulation mdr#mdd classification#mdd to new mdr classification#eu mdr#mdr medical#mdr regulation#eu medical device regulation#mdr classification rules#mdd classification rules
0 notes
Text
MDD to New MDR Classification of Medical Devices
The prerequisites for MDR classification for medical devices are basically equivalent to those in the present Medical Devices Directive (MDD). The EU MDR is shaking up the medical device industry and the order rules have not been left immaculate. The MDR decides the congruity assessment course for the device. While the order principally worries of the maker, if the device falls into Classes IIa, IIb, or III it has suggestions for the Notified body.
Before medical devices manufacturers can lawfully CE stamp their products in Europe, they should consent to the fitting medical devices order or guideline set out by the EU Commission. It is crucially critical to know the right medical device classification for your product before CE marking your devices. A classification impacts the administrative prerequisites for your devices, just as the endorsement course and its related expenses.
The number of rules in the MDR classifications has expanded from 18 to 22 and extra changes have been made to existing guidelines, which means numerous devices have new classifications. One of the important changes with the EU MDR is that medical device manufacturers will feel as they progress to conform to the new guideline is the adjustment in prerequisites for devices classifications. As medical devices classification change so do the prerequisites for manufacturers. The MDR will apply to specific products not directed through the MDD, such devices without an expected medical reason like non – corrective contact lenses.2 The MDR will likewise uncommonly manage devices joining nonmaterial’s and devices fabricated with non – feasible human tissue, which are at present absolved from the MDD.

With new guidelines likewise come classifications changes significant to specific devices. All dynamic implantable devices and their accessories will be considered as Class III. Any substance-based devices expected to be utilized by means of a body orifice or applied on the skin may not be a class I, so any substance – based devices at present in class I will be up – classified with the new regulation. Any manufacturer with a device that will be up – classified with the MDR must consent to the stricter necessities and will probably need to draw in their body more. The MDR classification specifies that the products are specifically intended for the cleaning, disinfection or sterilization of medical devices.
We at Operon Strategist help you in the process of making a defined and comprehensive technical file with all product details required for CE marking.
We’ll also provide assistance in your process of making technical file and review it at every step for compliance.
EU MDR classifications for medical devices
One of the first considerations for medical devices manufacturers who are seeking to place their products in the European markets is to determine which the appropriate classifications for their devices are.
The MDR classification of the device will impact on how and when you will engage with your Notified Body. As the market transitions from Medical Devices Directives (MDD) and the Active Implantable Medical Devices to the Medical Devices Regulation, the device manufacturers must note the changes in the requirements for device classification. For example, devices that had previously been included in the Active Implantable Medical Device Directives (MDD) are now covered in the Medical Device Regulation (MDR).
Medical Device Classification as per MDR
As per MDR Article, 51 Medical Devices are divided into class I, IIa, IIb and class III, taking into account the intended purpose of the devices and their inherent risks. Medical Device classifications are mainly based on the following factors:
Does the device have a standalone action?
How long the device is in continuous use in the human body?
Is it an invasive device or surgically invasive Medical Device?
Is it an implantable or active medical device
Does the Medical Device serve the purpose by the use of a certain drug?
The Medical Devices Classification in the European Union new MDR is outlined in ANNEX VIII, Chapter I and classification Rule mentioned in Chapter III. This chapter includes definitions of the terminology used in the medical devices guidance document for the classification of Medical Device.
The Medical Device is classified into:
Class 1
Class 1 Sterile
Class 1 Measuring
Class 1 Reusable
Class 11a
Class 11b
Class 111
EU MDR Medical Device Classification Rules
The Medical Device Classification EU rules, which are based on the vulnerability of the human body, should take into account the potential risks associated with the technical design and manufacture of the devices.
You’ll find all those rules on the Medical Device Regulation MDR 2017/745 Annex VIII
Rule 1– Non-invasive devices
Rule 2 – Non-invasive devices intended for channelling or storing (Which includes cells)
Rule 3 – Non-invasive devices that modify the biological or chemical composition of blood, body-liquids, other liquids and cells
Rule 4 – Non-invasive devices in contact with injured skin or mucous membrane
Rule 5 – Devices invasive in body orifices
Rule 6 – Surgically invasive devices for transient use
Rule 7 – Surgically invasive devices for short term use
Rule 8 – Surgically invasive devices for long term use and implantable (including any device administering medicinal products, surgical mesh or spinal disc)
Rule 9 – Active therapeutic devices intended to exchange or administer energy
Rule 10 – Active devices for diagnosis & monitoring, emit ionizing radiation
Rule 11 – Software intended to provide information which is used to take decisions with diagnosis or therapeutic purposes (from class I to class III)
Rule 12 – Active devices intended to administer and/or remove medicinal products, body-liquids or other substances
Rule 13 – All other active devices
Rule 14 – Devices incorporating a medicinal substance including human blood or plasma
Rule 15 – Contraception or prevention of the transmission of sexually transmitted diseases
Rule 16 – Specific disinfecting, cleaning and rinsing devices
Rule 17 – Devices specifically intended for recording of diagnostic images generated by X-ray radiation
Rule 18 – Devices utilizing non-viable tissues or cells of human origin or tissues of animal or derivatives.
Four new rules:
Rule 19 – Devices incorporating or consisting of nonmaterial
Rule 20 – Invasive devices with respect to body orifices to administer medicinal products by inhalation
Rule 21 – Substances or of combinations of substances that are intended to be introduced into the human body via a body orifice or applied to the skin and that are absorbed
Rule 22 – Active therapeutic devices with an integrated or incorporated diagnostic function which significantly determines the patient management.
Adjustments to the medical device classification system are not as problematic as those for IVDs yet will expect manufacturers to roll out some significant improvements. A large number of these progressions are a consequence of the old directives not considering the degree of the obtrusiveness and potential toxicity quality of certain devices. The MDR classifications of devices are into four classes: class I, class IIa, class IIb and class III. How they are classified relies upon 23 principles that think about their capacity, the risk to patients and the manufacturer’s intended use. There are a specific set of classification rules for four unique classes of medical devices: non-obtrusive, intrusive, dynamic and uncommon principles for inventive devices that incorporate different substances. Medical devices manufacturers ought to know that it is completely basic to classify these devices effectively from the earliest starting point as it directs the manufacturing prerequisites, clinical assessment and congruity evaluation. To ere in favour of alert, manufacturers should survey all present and future devices to guarantee consistence with the changed classification system. More info visit:https://www.operonstrategist.com/
#MDR classification#EU MDR classifications for medical devices#CE marking#Medical Devices Classification
0 notes
Text
CE marking for medical devices

As we all know CE Marking stands for CE stands for Conformité Européenne (French), which means European conformity. CE Marking focuses on Health and Safety & Environmental protection standard. The CE mark is compulsory conformity marking for specific products traded in the European Economic Area (EEA) since 1985. The CE Marking gives you intelligence to sell your product in the EU, Norway, Iceland, and Liechtenstein. The CE marking wipes out your need for interest in many competing jurisdictional laws covering your products. CE marking converts another level of safety for customers and other end-users, which lower damage claims and insurance premiums.
You must obtain CE marking on your medical devices which will give you the right to sell your medical devices in European countries (EU), CE marking determines that your medical device fulfills with the appropriate EU organizations and allows the commercialization of your products in 32 European countries. As a legal medical device manufacturer, you are responsible for managing regulatory assent and guarding CE Marking for your product, although of whether you outsource any or all components of your manufacturing operation. A manufacturer is described as a person who is able to design and manufacturing a product with the intention of placing it in on the market under their personal name or brand. Even a person really manufactures, designs, packs, assembles, labels or processes a product themselves, or they authorize out any or all of certain duties, does not imply in concerns to CE marking. The manufacturer has the responsibility to assure that the product and design are made in agreement with the relevant act. They must also pick up the expected professional documentation and assure those relevant assessment methods are carried out.
CE Marking logo affixed on the medical device shows the device fulfills the quality specifications and coordinated standards. Medical Device Regulation EU MDR Regulation (EU) 2017/745 and In Vitro Diagnostic Medical Devices Regulation (EU) 2017/746 are the two new regulations on the medical device.
Also, check ——>> CE marking certification in Sri Lanka
How to obtain CE marking for your medical device
CE is not a quality mark, but an understanding with EU Directives needs you to meet the appropriate standards of performance, quality, safety, and efficacy for your product type. However, the basic process follows these steps:
Determine which EU Directive applies to your device: Medical Devices Directive (93/42/EEC), In Vitro Diagnostic Devices Directive (98/79/EC) or Active Implantable Medical Devices Directive (90/385/EEC).
Determine the classification of your device. See our chart.
Classify all the EU directives and standard for the product applicant of CE marking.
Implement a Quality Management System, if applicable to your device. Most companies use ISO 13485 to meet the requirements.
Prepare a CE Marking Technical File or a Design Dossier.
Perform the conformity assessment according to relevant EU requirements of the directives.
Prepare a Clinical Evaluation Report (CER) according to MEDDEV 2.7/1 rev4 and MDD (or MDR).
Take necessary measures to make corrections of any divergence in the marking process.
Select and appoint a European Authorized Representative to act on your behalf within the EU if you have no physical location in Europe.
Attest the CE marking of the product.
Have your QMS and Technical File/Design Dossier audited by a Notified Body unless your device is Class I, is not sterile and has no measuring function.
Obtain CE Marking and ISO 13485 certificates from your Notified Body.
Arrange all the required documents and certificates.
Prepare and sign the EU declaration of Conformity.
Prepare a Declaration of Conformity (DoC), which states that your device complies with the appropriate Directive.
Also, check ——>> CE marking consultant in Sri Lanka
To make the CE Marking certification process more simple. You should hire a consultant when a consultant received your application they appoint a client manager who will guide you and your business through the following steps.
Gap analysis
Formal assessment
Certification and beyond
CE Marking & how to get Certified
NOTE: The process of the medical device will be changed when Europe’s new Medical Device Regulation (MDR 2017/745) comes into force in May 2020.
0 notes
Text
MDCG Guidance for Manufacturers of Class I Medical Devices

A medical device’s intended use and inherent risks must be considered when determining its MDR classification. Class I devices pose less risk to patients and end users, as under the previous MDD. The new Regulation EU MDR 2017/745 has added extended rules, leading some devices to fall under Class IIa, IIb, or even III.
This document is intended to guide Class I medical device manufacturers (excluding custom-made devices) that sell products bearing their name or trademark on the Union market to comply with MDR requirements. This guidance should also apply when an importer, distributor, or any other legal entity takes on the obligations owed by manufacturers.
The class I manufacturer must also hire a Notified Body (NB) to perform a conformity evaluation if medical devices are to be provided sterile, have measuring functions, or are reusable surgical instruments. For the NB to be qualified to conduct such a conformity assessment, the specific medical device in question must come within the purview of the Notified Body’s designation.
Large and mid-sized medical device manufacturers must also properly designate at least one individual in charge of regulatory compliance, but micro and small companies can function better by having a regulatory expert.
It is also important to designate an authorised representative residing within the EU if the manufacturer is registered outside of the EU. A foreign manufacturer must specifically issue a mandate outlining the representative’s duties and authority. It is also crucial to note that not all responsibilities associated with the medical equipment can be assigned.
The manufacturer must adequately preserve all the records required to verify compliance with the relevant requirements so they may be made available to the regulatory body upon request.
Steps to placing Class I medical devices on the market:
Manufacturers must ensure compliance with each of the following requirements if they plan to commercialise Class I medical devices. Please be aware that some of the outlined requirements are interconnected and can be completed in a different sequence than that which is shown.
The manufacturer will carry out a gap analysis for Class I devices that have already been released onto the market in compliance with the MDD to ensure that all of the below-listed requirements were satisfied at the time the MDR was applied.
The whole procedure includes a set of mandatory steps, including:
Step 0: Integrate MDR in the Quality Management System (QMS)
The manufacturer’s QMS should be appropriately linked with the standards outlined in the MDR. To ensure compliance with the following requirements, this will enable the correct assessment or decision to be taken and the appropriate documented evidence to be produced.
Step 1: Confirm product as a medical device
The intended purpose and the product’s mode of action are reviewed as per Article 2(1) of the MDR to ensure that the product qualifies as a medical device. If a product is assigned multiple intended purposes, it will qualify as a medical device only if all the intended purposes are covered in Article 2(1).
In the case of products identified as more than one category, the relevant legislation requirements will have to be followed. Despite not being medical devices in and of themselves, accessories to medical devices are subject to MDR regulations and qualify as devices under the MDR definition.
However, the MDR does not include accessories to devices covered by it as per Annex XVI of the MDR.
Step 2: Confirm product as a Class I medical device
The product has to be confirmed if it can be classified as a class I device as per Annex VIII of the MDR. Products previously classified as class I under MDD should be reviewed according to the classification rules of MDR to confirm if reclassification is required.
The MDD guideline cannot be used for devices that were moved from Class I to higher risk classes by the adoption of the MDR. The intended use of the device and any inherent risks related to the period of use, the part of the body, whether it is active or not, and whether it is invasive or non-invasive will determine how the classification standards are applied.
The classification rule with the highest class should be applied if the device in question falls under the purview of more than one classification rule due to its attributes.
Step 3: Procedures before placing on the market
a) Meet the General Safety and Performance Requirements (GSPR)
The devices will adhere to the general safety and performance standards outlined in Annex I of the MDR, considering the intended purposes that their manufacturers had provided.
The Risk management system is a continuous iterative process throughout the entire lifecycle of the product, established by the manufacturer that will enable the identification and analysis of the risks related to each device, the estimation and assessment of the risks associated with those risks, the elimination or control of residual risks, and the evaluation of the adopted measures based on the data gathered from the PMS system.
When a harmonised standard is in place, but a manufacturer chooses to use another reference, implementing that reference should, at the very least, ensure the same level of performance and safety.
Compliance with the relevant harmonised standards will give rise to a presumption that the MDR’s requirements, or portions of them, are also complied with.
If there are available standard specifications, the manufacturer must adhere to them unless they can adequately demonstrate that they have chosen a solution at least as effective and safe. The clinical evaluation processes, risk management, and PMS must be interdependent and updated regularly.
b) Conduct clinical evaluation
As part of the MDR’s technical documentation requirements, all devices—regardless of risk classification—require a clinical evaluation. The level of clinical evidence required to show compliance with the pertinent general safety and performance requirements listed in Annex I, which is obtained by considering the characteristics of the device and its intended purpose, shall be specified, and justified by the manufacturer.
Manufacturers must prepare, carry out, and record a clinical evaluation in compliance with Article 61 and Part A of Annex XIV to accomplish this.

Consideration of available alternative treatment options, the incorporation of clinical data and the acceptability of the benefit-risk ratio are required for carrying out a clinical evaluation.
Additional clinical data will be acquired or developed by clinical investigations if the currently available clinical data are insufficient to establish compliance with the MDR.
If the clinical data currently available for a device that is currently certified under Directive 93/42/EC is insufficient to show compliance with MDR, then post-market clinical follow-up studies of the device may be used to gather more clinical data.
Even data from the general post-market follow-up may occasionally be enough to close the difference.
a) Prepare technical documentation
The manufacturer is responsible for creating and maintaining the technical documentation to prove that their products adhere to the MDR’s technical specifications. Under Annex II and III, this technical documentation must be prepared before the EU declaration of compliance is drawn.
The manufacturer will develop and provide the technical documentation and, if applicable, it’s summary in a way that is unambiguous, clear, well-organised, and easy to search.
The manufacturer shall provide the CA, the authorised representative (where applicable), and NB with access to the technical documents (when applicable).
After reviewing the general safety and performance standards, as well as the pertinent technical provisions of the MDR, the technical documentation will be developed.
b) Request Notified Body involvement
Class I devices do not require the engagement of an NB for MDR compliance. But the manufacturer must follow the guidelines outlined in Chapters I and III of Annex IX or Part A of Annex XI of MDR when the product is a sterile equipment, measuring instrument, or reusable surgical tools.
For the relevant codes and corresponding types of devices as established by Regulation (EU) 2017/2185, manufacturers may select any NB designated in accordance with the MDR.
c) Prepare Instructions for Use and Labelling
Any safety and performance information required for a device’s safe usage and identification of the device should be provided along with the device by the manufacturer and/or the authorised representative, considering the possible users’ training and knowledge.
This data is included on the label, in the device packing, and in the instructions for use. Class I devices do not need instructions for use if they may be operated effectively and securely without them, deviating from the general rule.
Since Class Ir devices will need instructions for reprocessing (cleaning and sterilisation), an exception is most likely proposed.
Labelling and instructions for use must be written in accordance with national language regulations. There will be versions of the labelling and IFU in the technical documentation (in each pertinent national language).
The requirements regarding the information to be supplied with the device will be found in Annex I, Chapter III (23).
Step 4: Check compliance with general obligations for manufacturers
The manufacturer shall ensure that the general obligations for manufacturers outlined in Article 10 are met before releasing a device for sale.
Implementing a suitable QMS that will most effectively assure compliance with the MDR, such as through an internal audit, will receive special consideration. The QMS shall be documented, applied, maintained, updated regularly, and developed continuously, and it will at least include the following elements:
A strategy for regulatory compliance
Identification of applicable general safety and performance requirements and exploration of options to address those requirements
Responsibility of the management
Resource management, including selection and control of suppliers and sub-contractors
Risk management
Clinical evaluation, including post-market clinical follow-up (PMCF)
Product realisation, including planning, design, development, production and service provision
Verification of the UDI assignments
Setting up, implementation and maintenance of a post-market surveillance system
Handling communication with competent authorities, notified bodies, other economic operators, customers and/or other stakeholders
Processes for reporting serious incidents and field safety corrective actions in the context of vigilance
Management of corrective and preventive actions and verification of their effectiveness
Processes for monitoring and measurement of output, data analysis and product improvement.
According to applicable Union and national law, either natural or legal persons may seek compensation for harm brought on by a defective equipment.
In order to protect themselves against potential responsibility under Directive 85/374/EEC, manufacturers must take precautions that are commensurate to the risk class, type of device, and size of the business. These steps must not compromise further protective measures under national legislation.
Step 5: Draw-up the EU Declaration of Conformity
The process by which the manufacturer, who complies with the standards established by Article 52(7), certifies that the devices in question comply with the requirements of the MDR that apply to them is referred to in Article 19 as the EU declaration of conformity.
The CA will have access to the declaration of compliance, which must include, at a minimum, all of the data referred to in Annex IV. The manufacturer will regularly update the EU declaration of conformity and translate it into the official language(s) required by the Member States where the product is sold.
Suppose a device is subject to additional Union laws in addition to the MDR that also call for an EU declaration of conformity. In that case, the manufacturers will create a single EU declaration of conformity that refers to all applicable Union laws.
By creating the EU declaration of conformity, the manufacturer accepts liability for the device’s regulatory compliance with all relevant Union legislation.
For class Ir, Im, and Is devices, the manufacturer must obtain an EC certificate from NB per Annex IX, Chapters I and III, or Annex XI, Part A before applying a CE mark.
Step 6: Affix the CE marking
All Class I medical devices on the market must display the CE conformity label, which must be visible, readable, and permanent. It may be applied to the item or its sterile packaging. The CE marking must be applied to the package in cases where such affixing is impossible or unwarranted due to the nature of the device. The CE certification must be visible on all sales packaging and the instructions for use.
Placing the CE mark on a medical device signifies that it complies with all applicable safety and performance standards and is approved for marketing in the EU. The CE marking will be accompanied by the identification number of the relevant NB in the case of Class I medical devices placed on the market in a sterile condition, devices with measurement functions, and/or reusable surgical tools.
Affixing marks that could lead third parties astray about the significance of the CE marking is banned. Other extra markings may be applied to the product, the packaging, or the usage instructions, but they must not obscure or obstruct the CE marking.
The CE marking format will follow Annex V requirements. The minimum dimensions required for the CE mark may not apply to very small devices.
Step 7: Registration of devices and manufacturers in Eudamed
A streamlined approach might be used for medical devices that were previously put on the market per the Directives, provided that the manufacturer evaluates the gap and ensures that all regulations are properly followed.
A Class I medical device manufacturer must register the product with Eudamed before putting it on the market. Suppose the information listed in Section 1 of Part A of Annex VI has not already been registered in accordance with Article 31. In that case, the manufacturer must submit it to the electronic system in order to register the device. The information referred to in Section 1 of Part A of Annex VI will be given to that electronic system before applying to the NB in situations where the conformity assessment method necessitates the engagement of an NB in accordance with Article 52.
Following the CA’s validation of the manufacturer’s data in Eudamed, the manufacturer will receive an SRN from the aforementioned electronic system. To fulfil its duties under Article 29, the manufacturer will use the SRN when submitting an application to an NB for a conformity assessment and to gain access to Eudamed.
The registration of a device in Eudamed by the manufacturer includes:
Assigning a UDI-DI (with a Basic UDI-DI) as described in Part C of Annex VI to the device in accordance with the issuing entity’s policies mentioned in Article 27(2) and adding the UDI-DI (with a Basic UDI-DI) to the UDI database along with the other core data elements pertaining to that device mentioned in Part B of Annex VI.
Entering the data referred to in Section 2 of Part A of Annex VI, excluding Section 2.2 thereof, or, if previously provided, validating the data, and then keeping the data current up to date.
This document summarises the information presented above and outlines how Class I medical devices should be introduced to the EU market. The guidance’s scope also includes reusable surgical instruments (Class Ir), sterile medical devices (Class Is), and devices with measurement capabilities. These medical devices all need a Notified Body to be involved in pre-marketing procedures.
Originally Published at: https://omcmedical.com/mdcg-guidance-manufacturers-class-i-medical-devices/
0 notes
Text
MDD to MDR Transition for Medical Devices
The transition from the Medical Device Directive (MDD) to the Medical Device Regulation (MDR) marks a significant change in the regulatory framework for medical devices in the European Union (EU). The MDR came into full effect on May 26, 2021, replacing the MDD and introducing more stringent requirements for the marketing and oversight of medical devices.
Here are some key points to consider regarding the transition from MDD to MDR for medical devices:
1. Scope and Classification: The MDR has a broader scope and includes a wider range of medical devices than the MDD. It introduces new classification rules, which might result in the reclassification of some devices.
2. Conformity Assessment: The MDR emphasizes a risk-based approach to conformity assessment. It introduces stricter requirements for clinical evidence and post-market surveillance. Manufacturers need to provide more comprehensive data on safety, performance, and clinical benefit.
3. Unique Device Identification (UDI): The MDR mandates the use of Unique Device Identifiers (UDIs) for better traceability of devices throughout their lifecycle. UDIs provide information about the device's identity, origin, and production history.
4. Post-Market Surveillance: The MDR places greater emphasis on post-market surveillance and vigilance. Manufacturers are required to actively monitor the performance of their devices on the market and report any incidents or safety concerns.
5. Economic Operators: The MDR introduces new roles for economic operators in the supply chain, including importers and distributors. These entities have specific responsibilities related to device oversight and reporting.
6. Clinical Data Requirements: The MDR sets higher standards for clinical data and requires more extensive clinical evaluation for devices, especially for high-risk devices. Clinical data must support the device's safety and performance claims.
7. Notified Bodies: Notified Bodies play a crucial role in the conformity assessment process. The MDR requires stricter criteria for designation and oversight of Notified Bodies to ensure consistency and reliability in the assessment process.
8. Transitional Period: There was a transition period for manufacturers to comply with the MDR. Devices certified under the MDD could continue to be placed on the market until May 26, 2024, if the MDD certificate remained valid. However, for new devices, the MDR requirements are applied immediately after implementation.
9. Legacy Devices: After the transition period, devices that were certified under the MDD and placed on the market could still be used, provided they continue to meet their intended purpose and do not compromise patient safety. However, any modifications to these devices may trigger the need for MDR compliance.
It's important for manufacturers, importers, distributors, and other stakeholders in the medical device industry to understand the new requirements outlined in the MDR and ensure compliance to continue marketing and using medical devices in the EU market. The transition involves comprehensive changes, including adjustments to processes, documentation, and quality management systems to align with the new regulation. It's recommended to consult with legal experts and regulatory consultants to navigate the transition successfully.
IZiel team of specialists and quality professionals look forward to support more medical device companies to file their devices under the MDR 2017/745.
0 notes
Text
MDD to New MDR Classification of Medical Devices
#mdd to mdr#medical device regulation mdr#mdd classification#mdd to new mdr classification#eu mdr#mdr medical#mdr regulation#eu medical device regulation#mdr classification rules#mdd classification rules
0 notes
Text
Steps for Getting CE Marking
All products meeting the definition of medical devices as defined by the EU regulations require CE marking before they can be sold in the EU. CE marking indicates that a product has been assessed by the manufacturer and meets EU safety, health, and environmental protection requirements. It is required for products manufactured anywhere in the world that are then marketed in the EU.
The product must comply with all the relevant MDD to MDR requirements of before affixing the CE marking to it. Although the compliance requirements are similar in many ways, the European route is thought to be less governmental, resulting in shorter times to market, greater acceptance rates of new devices, and lesser costs associated with obtaining conformity certification.
While the benefits of obtaining a CE marking are significant, regulations are always subject to change. New modifications to the European medical device requirements are making the process more similar to that of the FDA when it comes to establishing conformity.
By May 2021, a new Medical Device Regulation (MDR 2017/745) will go into effect throughout the European Union. Despite the changes, there still remains a clear path to establishing conformity and obtaining a CE marking that will allow your company to access European markets.
Steps
1. IDENTIFY THE APPLICABLE REQUIREMENTS OF THE REGULATIONS
1. CLASSIFY THE DEVICE AS PER THE RULES DEFINED FOR CLASSIFICATION OF MEDICAL DEVICES
2. IDENTIFY AN APPROPRIATE ROUTE TO CONFORMITY
3. ASSESSMENT OF THE PRODUCT’S CONFORMITY TO EU REQUIREMENTS
4. COMPILE THE TECHNICAL DOCUMENTATION, QMS & CLINICAL EVALUATION REPORT.
5. ASSESSMENT BY NOTIFIED BODY
6. MAKE A DECLARATION OF CONFORMITY AND AFFIX THE CE MARK
IZiel Healthcare has collaborated with Belgium based Obelis (European Authorized Representatives) to provide a “One-Stop Solution” to fully support Class I, IIa, IIb & III medical device manufacturers across USA, Europe & Asia. Typically, all MDD-MDR transition projects initiate with Gap Assessment. Gap Assessment is a crucial activity during MDR transition.
IZiel-Obelis collaboration would ensure to obtain conformity with the MDR (2017/745) requirements and maintain the CE Marking by developing Technical File & QMS Documentation, conducting Software Validation, writing CERs and providing EC Rep, EUDAMED, PRRC Services. Our experts are well equipped to conduct this activity with an analytical mindset, resolve any engineering requirements and develop robust regulatory strategy for medical device manufacturers.
#regulation#ce marking certification#usfda#processvalidation#technical design document#software validation
1 note
·
View note
Text
Steps for Getting CE Marking
All products meeting the definition of medical devices as defined by the EU regulations require CE marking before they can be sold in the EU. CE marking indicates that a product has been assessed by the manufacturer and meets EU safety, health, and environmental protection requirements. It is required for products manufactured anywhere in the world that are then marketed in the EU.
The product must comply with all the relevant MDD to MDR requirements of before affixing the CE marking to it. Although the compliance requirements are similar in many ways, the European route is thought to be less governmental, resulting in shorter times to market, greater acceptance rates of new devices, and lesser costs associated with obtaining conformity certification.
While the benefits of obtaining a CE marking are significant, regulations are always subject to change. New modifications to the European medical device requirements are making the process more similar to that of the FDA when it comes to establishing conformity.
By May 2021, a new Medical Device Regulation (MDR 2017/745) will go into effect throughout the European Union. Despite the changes, there still remains a clear path to establishing conformity and obtaining a CE marking that will allow your company to access European markets.

Steps
1. IDENTIFY THE APPLICABLE REQUIREMENTS OF THE
2. REGULATIONSCLASSIFY THE DEVICE AS PER THE RULES DEFINED FOR CLASSIFICATION OF MEDICAL DEVICES
3. IDENTIFY AN APPROPRIATE ROUTE TO CONFORMITY
4. ASSESSMENT OF THE PRODUCT’S CONFORMITY TO EU REQUIREMENTS
5. COMPILE THE TECHNICAL DOCUMENTATION, QMS & CLINICAL EVALUATION REPORT.
6. ASSESSMENT BY NOTIFIED BODY
7. MAKE A DECLARATION OF CONFORMITY AND AFFIX THE CE MARK
IZiel Healthcare has collaborated with Belgium based Obelis (European Authorized Representatives) to provide a “One-Stop Solution” to fully support Class I, IIa, IIb & III medical device manufacturers across USA, Europe & Asia. Typically, all MDD-MDR transition projects initiate with Gap Assessment. Gap Assessment is a crucial activity during MDR transition.
IZiel-Obelis collaboration would ensure to obtain conformity with the MDR (2017/745) requirements and maintain the CE Marking by developing Technical File & QMS Documentation, conducting Software Validation, writing CERs and providing EC Rep, EUDAMED, PRRC Services. Our experts are well equipped to conduct this activity with an analytical mindset, resolve any engineering requirements and develop robust regulatory strategy for medical device manufacturers.
0 notes
Text
European CE Marking Strategy for Medical Devices
CE marking is very known requirement for those who are selling medical device in Europe. It is compulsory to have CE marking for all category of medical devices. This requirement is quite old and it was there even in MDD. However, with some additional new requirement in MDR, CE marking is slightly affected and one has to decide strategy for CE marking when transposing from MDD to MDR.
MDD also required CE marking and there is hardly any change in the approach but as we all know that CE marking is an indication that the device meets current EU requirement of medical device this automatically makes process strategized.
There are several changes happened from MDD to MDR which anyway all medical device manufacturer has to follow but CE marking process has not significantly but its requirement to meet current EU requirement makes all changes for manufacturers and marketers also.
Let us know simple steps where you can determine what needs to be done.
First decide which set of regulation applies to your device, Medical device regulation (MDR 1017/745) or IVDR (invitro diagnostic regulation) or (IVD1017/746)
Classification of your device as per MDR need to be reviewed based on risk assessment. Once the classification is complete one can know which regulation will apply.
Establish QMS. MDR requirement should be incorporated in your existing QMS with appropriate amendments or new QMS to be made for those who start now.
Create or review or renew your technical file to include all essential MDR requirement. This will also be a major change from MDD to MDR. In technical file review is very deep to check various dimensions including its appropriateness across its intended use, safety, labelling and packaging and effects of transportation and storage, risk v/s benefit for user.
For class I, IIa and IIb class technical file contains,
· Product description and specification
· Manufacturing information
· Risk management file
· Design verification and validation test reports
· Clinical evaluation
· Labelling
This process becomes smooth if QMS is strong.
For class III design dossier is an additional requirement which includes complete design process apart from data and technical file.
0 notes
Text
European Union Medical Device Directive (MDD) to Medical Device Regulations (MDR): strategic transition
This is especially important subject in medical device world. Understand strategic transition, one needs to go in detail as what is MDD with respect to MDR, why transition? And what transition?
MDD was labelled as directive, has quite relevant content but was not sufficient in view of rapidly changing technology which could deliver much advanced medical devices but also could have risk if it were regulated just by MDD. Objective of new MDR in broad category was to bring, improved consistency, better traceability, and transparency in regulatory process. Post market performance became particularly important aspect to judge newly designed advanced medical devices with modern technology.
It is especially important to note that MDR is regulation as ‘R’ of MDR stands for regulation and it is must to adopt. New deadline for MDR compliance was 26th May 2021. Now it is on and all those who are doing business of medical device in Europe has to see its compliance or else there will be huge implication on business.
Let us see major changes in this transition from MDD to MDR.
1. Product lifecycle approach like US FDA.
2. Device re-classification. If your already existing device falls into other high category of class, there will be tremendous implication on its management and monitoring.
3. The new updated Post Market Surveillance requirement.
4. General safety and performance requirement.
5. Clinical Evaluation requirement (CER)
6. Introduction of UDI (Unique device identification)
7. EUDAMED (EU databank on medical devices)
All above requirements are not just one time activity but must be covered under QMS and your existing QMS must be updated for risk management, clinical investigation/evaluation and post market surveillance.
These changes implies to medical device industry in terms of training of your all employees with new QMS and its execution so they can amend documented system in QMS.
As said above if device class is changed to higher level as per MDR reclassification, all requirements for new class then will be applicable. This may involve additional cost of compliance and may involve prohibitive cost to the same medical device which was already in market for long time at lower cost. This is big business challenge. If this new class calls for new clinical data, then it will be time consuming and costly too.
As the advancement in modern technology forced authority to transit MDD to MDR, manufacturer should also look for modern technology in their improvement too like in manufacturing, QMS. An automation is one of the best approaches to avoid human error and faster solution for implementation so one can be ready for both announced and unannounced audit.
Apart from transition steps mentioned above there are several existing process steps also need relook.
Process may need re-design with the support of automation for better consistency and no-defect. PMS process gains lot of importance so attention must be given to that. Success of this depends on your improved risk management process, product testing with new changed specification, gathering new clinical evidence. Supply chain compliances with integrated approach to have perfect traceability.
PSUR (periodic safety update report) is an important activity required under PMS system and management must device system under which this activity continues and meeting reviews, discusses and implements CAPA.
1. Serious incidents and field safety corrective actions from PMS.
2. Non serious incidents and undesirable side effects.
3. Trend data analysis.
4. Scientific or technical literature review.
5. Feedback and customer complaints.
6. Information about similar medical devices.
There is another report needed by MDR called PMSR (Post market surveillance report). Depending on class of your device one may need either PSUR or PMSR.
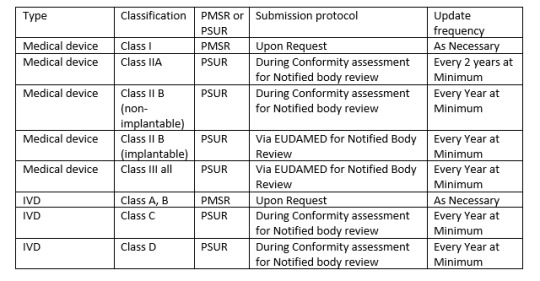
Since PSUR report is very essential, following items are essential for PSUR report,
1. PMS Data
2. Conclusion of Benefit risk analysis
3. Description of CAPA and rational or justification of the same.
4. Findings of PMCF (Post market clinical follow up)
5. Device sales volume and estimated user population.
6. Frequency of device usage (if practical)
7. An analysis and summary of all above listed items.
Similarly, PMS report is also needed to be regularly active activity. All manufacturers should capture data actively, analyse them and make results available to regulatory bodies.
Some companies offer software to manage all such activities so one cannot miss any such time bound activities and reporting the same to regulatory bodies.
Apart from above requirement it is obligatory on the part of manufacturer to ensure that, health and safety of user is not compromised due to defective, misleading, and dangerous products. When problem is detected, company should immediately take actions of Recall if needed. Develop a solution and keep users and regulatory body updated.
0 notes
Text
MDD to New MDR Classification of Medical Devices
#mdr device classification#mdd vs mdr classification rules#mdd vs mdr#mdr device classification rules#mdr classification#eu medical device classification rules#mdr classification rules annex viii#ce mdr classification#mdr classification examples#mdr risk classification#mdd to mdr changes
0 notes
Text

MDD to New MDR Classification of Medical Devices
The prerequisites for MDR classification for medical devices are basically equivalent to those in the present Medical Devices Directive (MDD).
Contact details –
Phone no - 93702 83428
Mail id – [email protected]
Visit- https://operonstrategist.com/mdd-to-new-mdr-classification/?utm_source=Image&utm_medium=Image&utm_campaign=Image
#mdr device classification#mdd vs mdr classification rules#mdd vs mdr#mdr device classification rules#mdr classification#eu medical device classification rules#mdr classification rules annex viii#ce mdr classification#mdr classification examples#mdr risk classification#mdd to mdr change
0 notes
Text

MDD to New MDR Classification of Medical Devices
The prerequisites for MDR classification for medical devices are basically equivalent to those in the present Medical Devices Directive (MDD).
Contact details –
Phone no - 93702 83428
Mail id – [email protected]
Visit- https://operonstrategist.com/mdd-to-new-mdr-classification/
0 notes
Text
MDD to MDR - Part 2 (How consultant can support you)
As old manufacturer of medical device you were naturally MDD compliant and to remain in business you will have to become MDR compliant too.
As Medical device business you also must have accounted the compliance cost to your product to assess the product cost and added your profitability to arrive at Product cost. The same exercise is now necessary as compliance cost will increase, in some cases substantially if your product class changes from say II to III. It is also difficult to explain to customer about increase in compliance cost may be some doctor may understand but overall, its difficult task. In view of this, whole exercise needs to be done with minimum expense and least possible time, so your approach must be thorough professional.
Taking clue from part I blog let us ask some questions to ourselves and get answer ready and actions if needed. Study of guidance document MEDDEV 2.7.1 is must.
1. Clinical evaluation now applies to all classes of product and this exercise is very big specifically for class III and product which will transit from Class II to Class III as per MDR. See our Blog on Clinical evaluation which describes in detail what is required so in terms of resource and cost, this is on top attention. This will no longer be just one time activity but life cycle continuous activity. Summary of safety and clinical performance (Article 32) stressing the need for Quality system on risk management. (Article 54 for class III and class IIb) General requirement of clinical investigation for conformity assessment (Article 63)
2. UDI system implementation is another big task and time consuming. Your resources must not only be enough but must also be adequately trained. (Article 27) This requires full system establishment each device will have unique device identification. The system should be electronic database so it can be made available to all required as mentioned in MDR.
3. Registration of device (Article 29)
4. Registration of Economic operator through electronic system. (Article 30) Single registration number (SRN) and procedure to communicate with commission or member state for SRN.
5. Registration of Manufacturer’s authorized representative and importers. (Article31)
6. Classification of device. Very essential and important documentation is needed here. This need to be maintained for each class of devices you are dealing with and how you arrived at specific classification using guideline points as per MDR. As mentioned in Part I classification of your existing device may change so entire technical documentation associated with that device. (Article 51)
7. Conformity assessment procedure (Article 52) and involvement of notified bodies (Article 53)
8. Eu Declaration of conformity (Article 19) The requirements specified in MDR is fulfilled for that specific device and technical documentation in each language is needed where this device is going to be marketed.
9. Requirement for a person of regulatory compliance (Article 15) and change of authorized representative (Article12) It is now mandatory to have a person on role with exclusive responsibility of regulatory compliance so naturally he will be completely knowledgeable person so additional resource and expense. Same is true for change of authorized representative where lots of documentation and transition period are defined.
10. Implant card and information to be supplied to patient. (Article 18) This is an additional compliance work.
11. post-market surveillance its plan and system (Article 84 and 83)
12. New reporting requirement of Periodic safety (Article 86), serious incident and field corrective action (Article 87), Trend data (Article 88). Analysis of serious incidents and field corrective actions (Article 89), Vigilance reporting is now within maximum 15 days.
13. Various annexures describing technical documentation need, a) conformity assessment based on QMS and assessment of technical documentation (Annex IX), b) conformity assessment based on type of examination (Annex X), c) conformity assessment based on verification (Annex XI), d) Clinical evaluation based on Post Market clinical follow up (PMCF) (Annex XIV), e) Clinical Investigation (Annex XV)
You have now many points to read and assess your system. Please ask one question to your team whether they understand all points well from MDR and guidance documents? There should not be any ambiguity or doubt or otherwise you will have non-compliance during audit. Your quality Management system must come under compliance. All actions decided during gap assessment must be complete. Review of that is needed before proceeding to next item. Attention now should be on Notified body. Check with team for readiness.
Please note that QMS is common for your organization but review your product portfolio and see whether you are ready for broader EU market? It is not just your organization, but your supplier and contractor also may pose challenge and their readiness. If not, you have mammoth task for changing them and bringing new.
Base on product portfolio make judge your readiness for certain product and wait for other till you are fully compliant. Gap assessment must also consider change of contractor if needed and impact of signed agreement. Micro and Macro management also need to be managed well to avoid unnecessary complexity and save time.
Do not forget training all people involved must undergo training to know new requirement to the extent they are supposed to know related to their responsibility. Knowing does not mean, just knowing the requirement but interpreting the same for your own product. This would need an external support where expert can pitch in.
Senior Managers expects that regulatory person knows well and can do what is needed. This is the area where what is planned and executed my not match an experienced third-party consultant like Iziel can identify the Gap which internal resource may not be able to do.
Where third party experts like IZiel can add value. Planning of entire Transition project and monitoring the same. Execution timings are essential for project timeline so expert consultant will prioritize activities such as longest time activity to start first where possible. Gap assessment identity and plan execution and Product portfolio understanding and rationalizing. Training
The complex project can be made easier by arming entire team with required knowledge of MDR articles and chapters along with required tools to apply.
Changes in clinical data will be another challenge as companies need to demonstrate clinical data with clinical claim and each intended use. Notified body will also be more vigilant on such aspects and will come prepared.
0 notes