#sbgrid
Explore tagged Tumblr posts
Visit Tumblr Blog
Explore Tumblr blogs with no restrictions, modern design and the best experience.
Last Seen Tumblr Blogs





Fun Fact
Tumblr posted its first advertisements in May 2012 and subsequently earned $13M in revenue.
Text
Mechanistic insight into odorant recognition
Our sense of smell is dependent on recognition of a vast array of odorants despite having a finite number of receptors. Odorants are mostly detected by G protein-coupled receptors called odorant receptors in olfactory sensory neurons. While there are only approximately 400 odorant receptors in humans, combinatorial activation of these odorant receptors enables sensing of odorants with diverse chemical structures. However, the mechanistic basis of odorant binding to odorant receptors in humans has remained unclear.
SBGrid member Aashish Manglik and other researchers have been working to develop a structural understanding of how odorants are recognized by odorant receptors. Using cryo-EM, they report the structure of active human odorant receptor OR51E2 bound to fatty acid propionate.
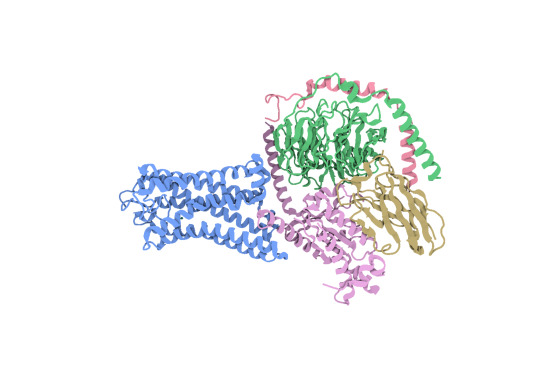
Above: Structure of human OR51E2 bound to propionate in complex with miniGs399. CC BY SBGrid.
Based on this structure, they determined that propionate is bound within a pocket in the odorant receptor and makes specific contacts to achieve activation. After mutating the odorant-binding pocket, they observed altered recognition of fatty acids with varied chain length. This suggests that activation of odorant receptors by odorants is influenced by tight packing interactions. Through molecular dynamics simulations, they show that propionate induces conformational changes in a specific region of odorant receptor OR51E2, extracellular loop 3. This work provides a foundational understanding of odorant recognition by human odorant receptors at a high-resolution, structural level.
Read more about this work in Nature.
#nature#science#cell#structural biology#sbgrid member research#sbgrid#biomedical research#cryo-EM#biomedicine#biomedical engineering
13 notes
·
View notes
Text
Ancient mediators of innate immunity
Bacteria can become infected by bacteriophages and have developed a range of anti-phage immune pathways to counteract these infections. These pathways are often multi-gene systems encoding proteins that sense and inhibit virion production, and efforts to catalog anti-phage signaling systems in bacteria have revealed that some of these genes share homology with components of eukaryotic immune systems. This suggests that eukaryotes horizontally acquired some innate immune genes from bacteria.
Many components have been identified as homologous between bacteria and humans, including bacterial cyclic-oligonucleotide-based anti-phage signaling systems (CBASS) with human cGAS and STING, and bacterial Viperins and Gasdermins with human Viperin and Gasdermin D. However, SBGrid member Aaron Whiteley and other researchers have been searching for other potential components in bacterial anti-phage signaling systems which could be homologous to immune signaling elements in humans. The researchers demonstrate that bacteria express anti-phage proteins containing a NACHT module, which is an important element of the animal nucleotide-binding domain leucine-rich repeat containing gene family called NLRs. These NACHT proteins are widespread in bacteria and contain a C-terminal sensor, central NACHT module, and N-terminal effector component, acting against both DNA and RNA bacteriophages.
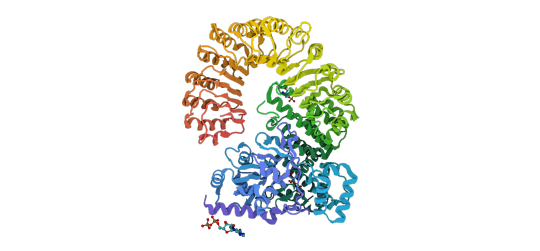
Above: Previously reported structure of NLR family CARD domain-containing protein 4. CC BY SBGrid.
Importantly, they determined that mutations in human NLR which lead to stimulus-independent activation of downstream signaling also activate bacterial NACHT proteins, suggesting that the bacterial and human systems share similar signaling mechanisms. This work identifies NACHT module-containing proteins as ancient innate immune signaling elements and expands our knowledge of homology between bacterial anti-phage immune pathways and eukaryotic immune systems.
Read more about this work in Cell.
#cell#immunology#science#nature#microbiology#sbgrid#sbgrid member research#biomedical research#Biomedical Science
7 notes
·
View notes
Text
Understanding how iron affects stearoyl-CoA desaturase-1 activity.
Enzymes are the manufacturing facilities of the cell. They take the materials provided by the environment and convert them into cellularly relevant chemicals. The physical processes that allow enzymes to do this vital work are of interest to scientists because of the possibility of treating enzyme-related diseases, using enzymes as therapies for neurodegenerative diseases, and harnessing their manufacturing process for production of chemicals relevant to society. In a recent study by SBGrid member Ming Zhou from Baylor College of Medicine, the maintenance and function of a subclass of enzymes becomes more apparent.
Stearoyl-CoA desaturase-1 (SCD1) is an enzyme that localizes in the endoplasmic reticulum of cells and contains two iron atoms at its core. SCD1 uses the core irons to carry out the catalytic function of adding a double bond to fatty acids, thus creating unsaturated fatty acids. This enzyme contributes to diseases like cancer and diabetes and is a possible target to treat neurodegenerative diseases. Zhou and colleagues pursue an answer to how and why SCD1 loses enzymatic activity after a certain amount of uses.

Above: Published structure of SCD-1 diiron active site. PDB: 6WF2. CC BY SBGRID
Similar to common machines in a household, enzymes stop functioning after a certain number of uses. Understanding why a machine stops working can provide insight about how to keep them working longer and more effectively. Zhou and company used enzyme activity assays to show that SCD1 lost function after 8.5 reactions due to the loss of an iron from the active site of the enzyme. The team used UV-vis spectroscopy, EPR, and inductively coupled plasma mass spectrometry (ICP-MS) to confirm that the loss of activity was due to the loss of iron in the active site. They also found that this iron loss could be remedied by providing an excess of available iron for SCD1 to take up and continue its work, though once SCD1 becomes inactive this treatment can not restore activity. This provides a possible “repair” for low SCD1 activity by ensuring that it has a large pool of possible iron molecules with which to maintain its function. This study provides interesting insight into how to limit and possibly prevent enzyme self-inactivation.
Read more about this work in the Journal of Biological Chemistry.
-Vida Storm Robertson, Fisk University
4 notes
·
View notes
Text
A new method for HIV antibody design
Human Immunodeficiency Virus (HIV) is a highly contagious virus that attacks and breaks down a person’s immune system. If left untreated, HIV can develop into acquired immunodeficiency syndrome (AIDS). Although there is no cure for HIV or AIDS, there are treatment options that can reduce the detectable viral load in HIV patients and prevent the progression into AIDS. There are also HIV vaccines and other therapeutics that aim to reduce the rates of viral transmission in humans. Broadly neutralizing antibodies (bNAbs) are a class of antibodies that are potent and can neutralize many strains of HIV and have been used in HIV vaccine design. However, higher potency and improving the breadth of neutralization for HIV strains is needed to make bNAbs more effective at limiting viral transmission.

Above: Cyro-EM structure of PG9RSH Y(100k)D variant. CC BY SBGRID.
SBGrid member Peter Kwong, and colleagues, were able to create three bNAb variants with improved potency and neutralization breadth using their own computational protein design software, OSPREY(open-source protein redesign for you) and tested them, along with their wildtype counterparts and a best-in-class antibody, against 208 pseudo viruses. They found that their newly designed bNAb variants showed improvements in potency and breadth as compared to the wildtype and best-in-class antibodies. They were also able to determine cryo-EM structures of the three variants to further study how the observed increases in potency and breadth were achieved from a structural viewpoint.
Read more in Cell Reports.
-KeAndreya Morrison, Meharry Medical College
2 notes
·
View notes
Text
New structure of flagellar motor component “MS-ring” uncovers details about changing stoichiometry and conformation
How bacteria move around the body and cause infection has been a topic of debate among scientists for years. One way bacteria know where to go is through a special sensing mechanism that allows bacteria to adapt and move around their environment is called chemotaxis. Chemotaxis is the ability of organisms to follow a concentration gradient in response to a changing environment around it. A key structure involved in chemotaxis in bacteria is the flagellum, a squiggly tail that protrudes from the end of a bacteria and helps propel it forward. The importance of chemotaxis and how the flagellum moves has led many to look to the structure of a particular piece of the flagellum known as the MS-ring, which is a central component of the flagellar motor. Although there are a few reported structures of the MS-ring, these reports conflict about which and how many domains actually comprise the MS-ring.
SBGrid members Tina Iverson and Terunaga Nakagawa, with Prashant Singh, have been working to elucidate a cryoEM structure of the MS-ring and uncover the structural basis for the apparent flexibility in stoichiometry and domain placement. By co-expressing multiple domains of the MS-ring and another component of the flagellar motor, the C-ring, the authors were able to study a “post assembly” complex of the MS-ring and examine the stoichiometry of the MS-ring domains. They found that the MS-ring can adopt more than one stoichiometry. The authors did suggest that differences in stoichiometry in conflicting reports could be explained by different growth conditions during expression. However, it was also stated that this difference could be a result of the flagellum increasing its torque, by increasing certain domains within the MS-ring.

Above: Top view (left) and side view (right) of the post assembly structure of the MS-ring. CC BY SBGrid.
The structure observed here, and the comparisons made between the previously reported structures, show that structural plasticity, or flexibility, happens on many levels within the MS-ring of the flagellum. The biological reason for this phenomenon could be how the flagellum responds to chemotaxis and changes in the environment.
Read more about this work in PLOS ONE.
-KeAndreya Morrison, Meharry Medical College
3 notes
·
View notes
Text
Structural adaptations underlying distinct sensing behavior in octopus and squid
Octopus and squid are coleoid cephalopods with large nervous systems and flexible arms which are capable of advanced sensing and processing. Both squid and octopus use cephalopod-specific chemotactile receptors (CRs) to sense in their environment. In octopus, gene duplication and structural adaptations of the ancestral nicotinic acetylcholine receptors enabled the development of specialized CRs capable of contact-dependent chemosensation of ligands that are poorly soluble, supporting their extensive foraging behavior. In comparison, squids are characteristically ambush predators.
SBGrid member Ryan Hibbs and other researchers have been working to elucidate the connection between the divergence in the CR structure in squid and octopus and the evolution of lineage-specific functionality to sense molecules suited for specific physiological roles. They determined that while squid express ancient CRs more comparable to nicotinic acetylcholine receptors, octopuses express a different, more recently developed form of CRs which enables their “taste by touch” sensing. The authors report the structure of the squid CR and compare it with the octopus CRs and nicotinic receptors, revealing evolutionary structural adaptations in the octopus CRs. Specifically, the ancestral aromatic cage coordinating recognition of soluble ligands transitioned to a hydrophobic binding pocket that traps insoluble molecules to enable octopus CR-mediated contact-dependent chemosensation.

Above: Structure of squid sensory receptor CRB1. CC BY SBGrid.
This work demonstrates how the modification and expansion of sensing functionality enabling specific behaviors can be traced to divergence in protein structure.
Read more about this work in Nature.
3 notes
·
View notes
Text
Using flies to study neurodegeneration
This publication highlight is part of the SBGrid/Meharry Medical College Communities Project, focused on science education and demonstrating how structural biology and preclinical science connect to medicine.
In the body, proteins carry out vital tasks to keep us alive. In order to do the wide variety of tasks needed to keep a living system going, proteins can form unique structures in order to carry out each unique function. These structures are encoded into the protein by its amino acid sequences, which are in turn determined by an mRNA sequence that is encoded by a DNA sequence. Information flows from DNA to RNA to protein structure in the cell and if any problems occur in this information pipeline, detrimental consequences follow. One example of a problem in the pipeline is a mutation in the DNA causing proteins to fold into the wrong shape and not be able to carry out their function. This is the case for Huntington’s Disease. In this neurodegenerative disease, the DNA that makes up the Huntington gene is mutated, almost resembling a skipping record, repeating the same nucleotide phrase, cytosine–adenine–guanine (CAG), over and over again. This repetition causes the resulting protein to have long regions unfolded or misfolded because of repeats of the glutamine amino acid (Gln, Q) and is referred to as a polyQ disease. Proteins with polyQ tracts are enriched in genes with neuronal function and typically aggregate together and cause the tragic symptoms that are hallmarks of the disease. In a recent publication in a IUCr Journal, SBGrid member Dr. Dierk Niessing, publishes a new protein structure that could help limit the aggregation of Huntington gene proteins.
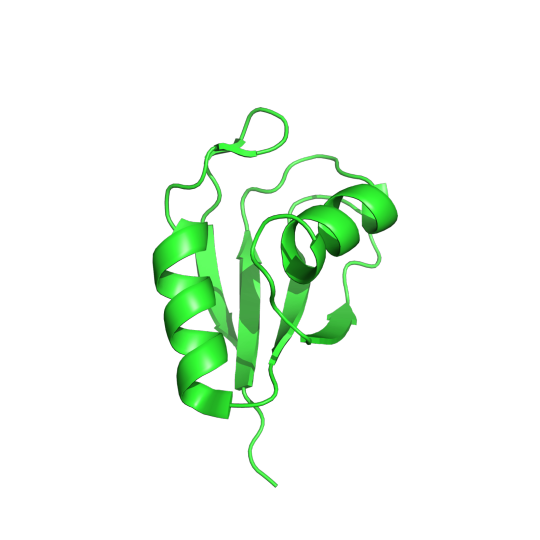
Pictured above is the determined structure of TRMT2A in Drosophila melanogaster. PDB:7PV5. CC BY SBGRID
Recent studies have shown that the inhibition of a protein called TRMT2A can lessen the aggregation of Huntington proteins, which means that TRMT2A could be a potential target for therapeutic molecules. If a drug can inhibit TRMT2A activity, it has the potential to help with Huntington’s disease. In order to determine how well this hypothesis works, we need a model organism that allows for ethical experimentation. Luckily, Dr. Dierk Niessing found a homolog of TRMT2A in the common fruit fly. He used protein crystallography and x-ray diffraction to observe the structure of this homologous protein. Upon examination, this fruit fly homolog had a different sequence than the human TRMT2A protein but a very similar structure. All the catalytic residues, the functionally relevant amino acids in the human protein, were also conserved in the fruit fly protein. This paper builds a case using structural biology that this fruit fly protein is a very similar homolog to the human TRMT2A protein. The implications of these findings mean that fruit flies could be an effective model to study how well drugs that target this protein are able to lessen Huntington symptoms. Read more about this fruit fly homolog in the Acta Crystallographica Section F.
-Vida Storm Robertson, Fisk University
Vida Storm Robertson is a Masters Student in Chemistry at Fisk University working in both solid-state and solution based structural determination techniques. He plans on starting a PhD program in biophysics in the fall of 2024.
0 notes
Text
Metals as nutrients for bacteria to thrive
This publication highlight is part of the SBGrid/Meharry Medical College Communities Project, focused on science education and demonstrating how structural biology and preclinical science connect to medicine.
Copper is a micronutrient that helps regulate many important functions needed for bacteria to survive. However, high amounts of copper can be toxic to bacteria so it’s important for bacteria to be able to maintain homeostasis. Copper’s main role is to act as a cofactor. A cofactor is a non-protein molecule that is crucial for a protein to carry out its function. Many proteins use copper as a molecular helper; because of this, copper is essential for all living things, yet, it is still unclear how bacteria get and maintain their copper levels. The work titled Stabilization of a Cu-binding site by a highly conserved tryptophan residue by SBGrid member Oriana Fisher looks into how the Gram-positive bacterium Bacillus subtilis regulates copper levels.
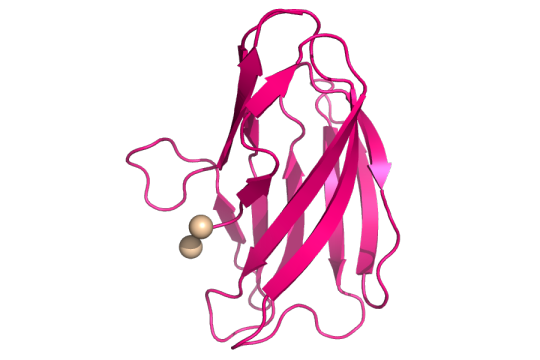
Structure of copper(II) (tan spheres) bound to the mutant YcnIW137F. CC BY SBGRID.
The authors looked at the ycnKJI operon that contains three copper-using proteins. An operon is a group of genes that are all controlled by the same promoter, or the place where genes are turned “on”. They found that a member of the ycnKJI operon, YcnI, a copper-binding protein whose function is still unknown, prefers to bind the oxidized version of copper. This oxidized version is known as Copper(II) oxide or Cu(II). By creating a mutated version of Ycnl where they swapped out a conserved tryptophan for phenylalanine, they found that even though tryptophan, an amino acid and binding site of Cu(II), is found in about 98% of this family of copper-binding proteins, it is not essential for Cu(II) binding. The conserved tryptophan, however, is necessary for maintaining the stability of Cu(II) binding to the protein.
Read more in the Journal of Inorganic Biochemistry.
- KeAndreya Morrison, Meharry Medical College

0 notes
Text
A small molecule that clogs up ribosome machinery
This publication highlight is part of the SBGrid/Meharry Medical College Communities Project, focused on science education and demonstrating how structural biology and preclinical science connect to medicine.
Ribosomes are essential molecular machines in the cell. They create proteins through the translation of RNA into an amino acid sequence. This process of translation is an important topic of study for molecular and structural biologists because of its vital role in maintaining life and the proteome. The production of the full proteome is essential for life and any variance or interference in the process could lead to drastic consequences. Because of this, the study of how small molecules and drugs interact and influence the ribosome is an ever-present question for structural biologists and medicinal chemists. In Nature Chemical Biology, SBGrid member Dr. Susan Shao from Harvard University, shows the unique effect that a small molecule, SRI-41315, has on the ribosome.
Ribosomes are made of many different protein and RNA units. The process of creating amino acid chains from an RNA transcript is highly dynamic and requires the timely introduction and removal of a library of participating molecules in the translation symphony. One of these molecular players in translation is eukaryotic translation termination factor 1, eRF1. This protein binds to the ribosome subunits and aids in the termination or conclusion of the translation process. eRF1 helps the ribosome read the translation instruction, the mRNA, and helps the ribosome know when to stop and remove the finished amino acid chain. The small molecule SRI-41315 has been known to degrade eRF1, but the mechanism for this degradation was unknown until Dr. Susan Shao and colleagues proposed the following using cryo-EM and molecular assays:
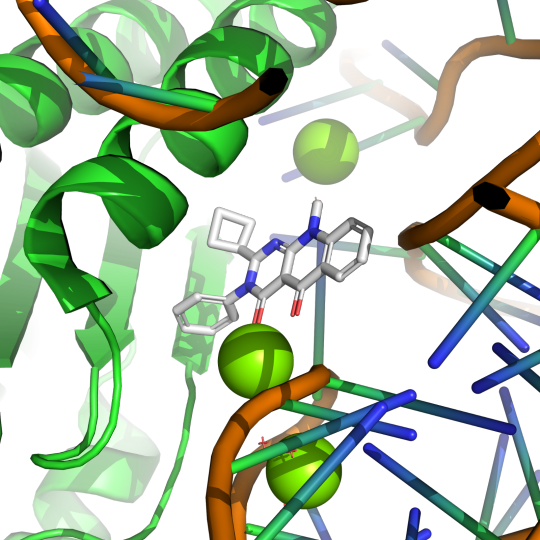
Pictured above is small molecule SRI-41315 (gray) interaction with eRF1 (green) and RNA (orange) in the ribosomal complex. PDB:8SCB. CC BY SBGRID
Using electron microscopy at cryogenic temperatures, a technique known as cryo-EM, Dr. Shao was able to create a model of eRF1 in complex with a ribosome with SRI-41315 bound. In this model, SRI-41315 was shown to act as a glue in the machine. This small molecule locked eRF1 into a specific configuration, limiting the dynamic process of translation. By gumming up the ribosomal machine, SRI-41315 increased eRF1 degradation and affected the machine's ability to accurately produce functional proteins. The ability of this small molecule to indirectly slow down and effect the efficiency of translation is a very compelling observation and provides evidence for the wide array of functions small molecules can have in living systems.
To learn more about the clog in the ribosomal machine, continue reading at Nature Chemical Biology.
Vida Storm Robertson, Fisk University
Vida Storm Robertson is a Masters Student in Chemistry at Fisk University working in both solid-state and solution based structural determination techniques. He plans on starting a PhD program in biophysics in the fall of 2024.
1 note
·
View note
Text
An inside look into the Powerhouse of the Cell
This publication highlight is part of the SBGrid/Meharry Medical College Communities Project, focused on science education and demonstrating how structural biology and preclinical science connect to medicine.
Mitochondria are the powerhouses of the cell. We’ve all heard this phrase at some point in our lives, but what does it mean on a molecular level? A mitochondrion is an organelle in cells like animals, plants and humans that functions to provide the cell with energy in the form of a molecule called adenosine triphosphate, or ATP. A sufficient stock of ATP is produced in mitochondria when cells in our body convert food into energy, or what is known as cellular respiration. More specifically, cellular respiration is when our cells take in glucose and oxygen through many steps and then release carbon dioxide, water, and energy. An important step is to store this energy into a form that can be used in different parts of the cell when needed. This is done by moving electrons (really small particles that are a part of atoms) by a group of biomolecules called proteins. These proteins come together to form an electron transport chain. As the name suggests, the electron transport chain is a series of processes where electrons are transported from one molecule (donor) to another molecule (acceptor) resulting in the formation of ATP. For the electron transport chain to work, it relies on electron donor molecules produced from the Krebs cycle, also known as the citric acid cycle.
The Krebs cycle is a metabolic pathway in the mitochondria that allows our bodies to break down carbohydrates to be used for energy. This process starts with pyruvate entering the system and, after several steps, ends with the production of several molecules such as, ATP, NADH, and FADH2. These important molecules enter the electron transport chain acting as electron donors to make more ATP. CO2 is also produced from the Krebs cycle and enters other metabolic pathways. All of the steps in the Krebs cycle are carried out by different enzymes. One of the enzymes is succinate dehydrogenase (SDH). As a member of the electron transport chain in mitochondria, SDH or complex II, catalyzes the formation of fumarate from succinate- releasing FADH2 as a byproduct. SDH produces a fraction of the total ATP, but is important for a wide range of cellular activities, such as suppressing tumor growth and neurological development. Although the importance of SDH has been widely established, what is unknown are the details of how the regulation of SDH assembly factors (SDHAF) and SDH assemble into a functional unit.
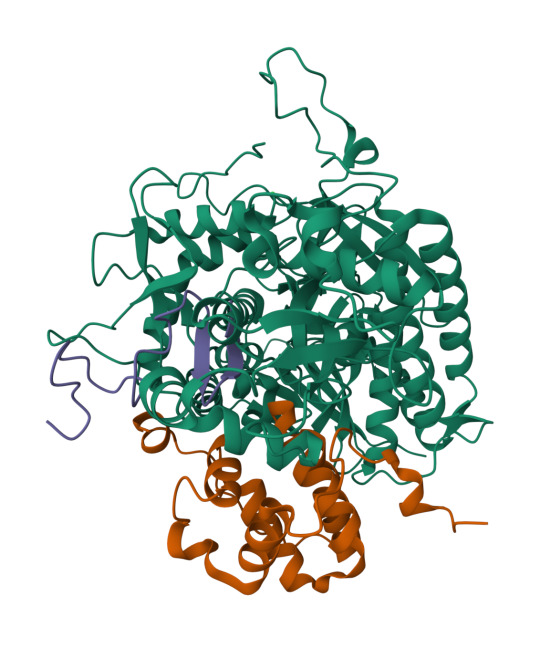
Left: structure of SDHA-SDHAF2-SDHAF4 assembly intermediate (PDB: 8DYD). Green: SDH, purple: SDHAF4, orange: SDHAF2. Right: structure of SDHA-SDHAF4 assembly intermediate (PDB: 8DYE). Green: SDH, orange: SDHAF4. CC BY SBGRID.
To address this question, SBGrid member Tina Iverson investigated the intermediate complexes formed by the SDHAF subunits and cofactors. Through the use of cellular studies and biochemical experiments, along with X-ray crystallography and NMR for structural studies, the authors were able to map out the different intermediate complexes that lead to the formation of the fully matured SDH. Interestingly, the authors found that regions of the assembly proteins can transition from ordered (a protein with a secondary structure) to disordered (a protein with no secondary structure) and that this change is important for the transfer between intermediate complexes. These results shed light on the previously unknown steps of the formation of a mature and functional complex II of the electron transport chain.
Read more in Nature Communications.
KeAndreya Morrison, Meharry Medical College

0 notes
Text
Do you know what can cause acne?
This publication highlight is part of the SBGrid/Meharry Medical College Communities Project, focused on science education and demonstrating how structural biology and preclinical science connect to medicine.
Everyone will have some experience with acne in their lifetime. Generally, most people struggle with acne during their teenage and young adult years, but acne can persist in some people during their adult years. Acne is a condition where pores and hair follicles in the skin get clogged. Pores can be clogged with dead skin cells, sebum (an oily substance that provides a barrier for the skin), and bacteria. Clogged pores create pimples that can swell and become painful. Cutibacterium acnes (C. acnes) is a common bacterium that plays a role in the health of our skin . It is the bacteria most commonly associated with acne even though it is found on healthy and acne-prone skin similarly. Like many other bacteria, C. acnes has multiple strains. Some strains are associated with acne-prone skin while others are associated with healthy skin. However, not much is known about the genetic factors that make a strain of C. acnes acne-causing. One key difference between C. acnes strains that promote healthy is the variation in the enzyme hyaluronidase. There are several television commercials that mention acne treatments that contain hyaluronidase. Hyaluronidase is an enzyme that breaks down hyaluronic acid. C. acnes can express two variants of hyaluronidases, HylA and HylB. HylA and HylB are about 74% similar. HylA is associated with acne-prone skin while Hyl B is associated with healthy skin.
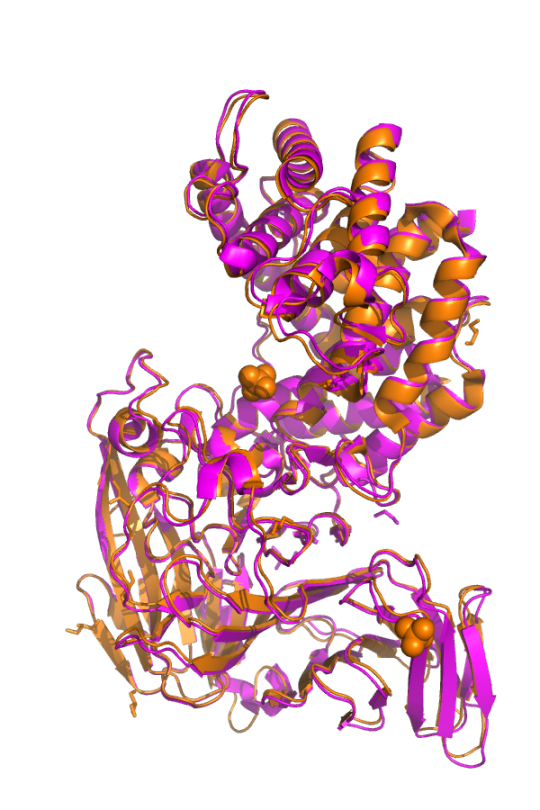
Pictured above is HylA (pink; PDB: 8FYG) overlayed with HylB (orange; PDB: 8FNX). CC BY SBGRID.
In this work, titled Functional divergence of a bacterial enzyme promotes healthy or acneic skin, SBGrid member Ramachandran Murali and colleagues uncover the different mechanisms of action between HylA and HylB that contribute to acne or healthy skin, respectively. HylA breaks down hyaluronic acid into large fragments. These large fragments help promote an inflammatory pathology that leads to acne. Contrastingly, HylB degrades hyaluronic acid solely into disaccharides (two sugar molecules) that lead to reduced acne. Along with solving x-ray crystal structures of different mutants of HylA and the wildtype of HylB, the authors also show the potential for targeting HylA using inhibitors and peptide vaccines. Their findings suggest that inhibiting HylA could be a potential target for future acne treatments.
Read more in Nature Communications.
- KeAndreya Morrison, Meharry Medical College

0 notes
Text
New SARS-CoV-2 antibody generated to bind to multiple conformational states of spike proteins.
This publication highlight is part of the SBGrid/Meharry Medical College Communities Project, focused on science education and demonstrating how structural biology and preclinical science connect to medicine.
The emergence of SARS-CoV-2 in 2019 created a worldwide pandemic that is still present in the population today. Scientists like Dr. Jason McLellan and Dr. Kizzmekia Corbett have published methods for isolating stable versions of SARS-CoV-2 spike proteins that can be studied in a research lab. Structural biologists and pharmacologists have created many effective treatments and vaccines for this respiratory virus, but the efficiency of these therapies changes as the virus mutates. The high viral load of SARS-CoV-2 in the population allows for constant evolution of the virus, specifically selecting for mutates able to avoid prevalent treatments. Thus, it is no surprise that the latest variant, Omicron, showed a unique ability to avoid immune detection and recognition from formerly designed antibodies. Drs. Jason McLellan and Andrew Ellington, in their latest paper in Communication Biology, show a unique method of developing new antibodies and describe how this newly developed antibody performs against the most prevalent variants of SARS-CoV-2.

Pictured above is the receptor binding domain of COVID-19 (green) binding to antibody N3-1 (blue). PDB:6M0J. CC BY SBGRID
New variants of SARS-CoV-2 are able to avoid the immune system's detection and current antibody binding because of mutation and variations in dynamics. To maintain the effectiveness of current COVID-19 treatments new antibodies must be designed to bind to these emerging mutates. In this paper, new antibodies that preferentially bind to the spike protein of new SARS-CoV-2 mutates are designed using an integrative cellular and structural approach. Antibodies are made up of two interchangeable parts, a heavy chain and a light chain. The heavy chains of antibodies created in this paper were taken from serum donated by patients who had previously contracted COVID-19. These patient-derived heavy chains were combined with light chains that were previously published in public data banks. Combinations of heavy and light chains were tested for efficiency of binding to SARS-CoV-2 spike protein. A novel antibody created using the methodology above showed high efficiency. This unique antibody's high binding was due to its ability to bind to many different conformations of the spike protein. This ability to bind to the SARS-CoV-2 spike protein, in spite of its protein dynamics, allows this new antibody to be much more effective at targeting more evasive strands of the virus.
To read more about SARS-CoV-2 and this new method of antibody development, check out the paper in Communications Biology.
- Vida Storm Robertson, Fisk University
Vida Storm Robertson is a Masters Student in Chemistry at Fisk University working in both solid-state and solution based structural determination techniques. He plans on starting a PhD program in biophysics in the fall of 2024.
0 notes
Text
Structure of Equity - Jamaine Davis - Meharry Medical College
Sharing some of our #SBGrid member tales from the last year. This one from September 2022.
Numbers speak clearly to Jamaine Davis. As a boy growing up on Long Island, math came so easy to him that one of his family nicknames was "the professor."

Other numbers have shaped his ambitions at Meharry Medical College in Nashville, Tennessee, where Davis runs one of the few labs in the world that uses structural biology to help explain biological health disparities.
For example, U.S. Black adults are twice as likely to have Alzheimer's disease compared to non-Hispanic Whites. And despite a somewhat lower overall lifetime risk of breast cancer, Black women experience a 40% higher death rate from breast cancer than White women at every age and are more likely to be diagnosed with fast growing and late-stage breast cancer.
"My research program is basically at the intersection of structural biology, genetics and disease, and health disparities," says Davis of the big-picture questions that guide his lab's work. "What are the molecular mechanisms that dictate who develops diseases like cancer or Alzheimer's? And then how do we design effective therapies? How do we target the right pathways for the right treatment for that patient?"
One project in the early stages focuses on a gene (ABACA7) that has a stronger effect on risk of Alzheimer's disease in Blacks than the better known ApoE4 gene risk variant. "It's actually the strongest risk factor for developing Alzheimer's in African Americans known so far," Davis says.
As he explains it, ABACA7 transports lipids out of cells, handing off the lipids directly to ApoE, and also interacts with Tau, another protein that goes awry in Alzheimer's. Two missense variants in ABACA7 confer the risk.
"So we've been studying these mutations to see what impact they have on lipid transport," Davis says. "Once we're done, we can look at the people who particularly carry this mutation or variant, see what downstream processes are altered, and design therapies to rescue that. And these variants so far have only been identified in African Americans."
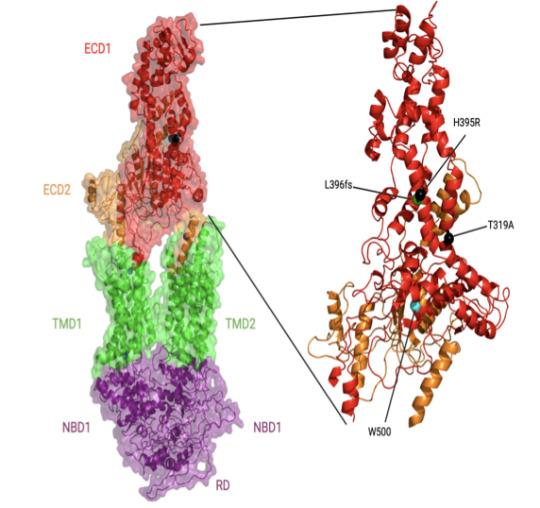
In individuals with African ancestry, the phospholipid-transporting ATPase ABCA7 (ABCA7) gene has stronger associations with Alzheimer’s disease risk than in individuals with European ancestry and than the apolipoprotein E (APOE) ɛ4 allele. The Davis lab is exploring the structure and function of key ABACA7 mutations and how they contribute to alterations in transporting lipids, which may influence Alzheimer’s in African Americans. Credit: Courtesy of J.Davis.
Davis began his academic training on a different career path. With his early affinity for math and science, he reasoned that chemical engineering made sense as a college major. But near graduation at Drexel University, he realized that the typical next step for someone with a chemical engineering degree was a job at an oil company. He hadn't taken one biology course in college, but he found himself drawn to biomedical research instead.
He seized an opportunity to work in a biophysics lab at a neighboring school, University of Pennsylvania, where his mentor Jacqueline Tanaka gave him a peek at the scientific career he could have in biophysics and opened his eyes to the kind of academic role model he could be. Her excitement for X-ray crystallography and for increasing the proportion of women and minorities in science inspired him to go to graduate school.
"She built her career in structural biology and mentoring, hand in hand," Davis says. "She saw some potential in me, and I was at a crossroads." Davis had also been unaware of the extent of health inequities across the country and of the low representation of minorities in academia.
For his thesis, Davis chose the lab of Harvey Rubin, a dynamic speaker who fostered an immediate interest in infectious disease. In Rubin's lab, Davis characterized an enzyme that enables Mycobacterium tuberculosis to enter (and possibly exit) the dormancy stage in the lungs of people.
When Davis finished his PhD in 2007, he was the first Black to earn a doctorate in biochemistry and molecular biophysics at UPenn. "I had a great time," he says. "They were very supportive. But it is pretty shocking. If you look at Twitter, there are other people posting the same kind of statistic. They're the first Black to graduate from a certain program at a certain institution. It does show there is still some under-representation across different departments."
He followed up with two postdoctoral fellowships at the National Cancer Institute. He first showed that a novel protein in Shigella (bacteria that cause food poisoning) was not a protease, as some suspected. A second project elucidated the binding modes of a protein with multiple domain repeats implicated in the development of cancer.
Then he thought about how best to combine his interests in a distinctive research program. He chose Meharry, one of the oldest and largest historically black U.S. academic health centers. (Davis is also a member of the Vanderbilt University Center for Structural Biology.)
Historically black colleges and universities are powerhouses in educating African Americans who go on to earn doctoral degrees in science, technology, engineering, math, and medicine, as Davis and his co-authors reviewed in a commentary (Cell, 2022). Blacks make up 12% of the U.S. workforce, but only 5% of working physicians and 3.6% of full-time faculty conducting research at medical schools.
When the COVID-19 pandemic hit, Davis found new opportunities for mentorship and community outreach. Soon after the pandemic took hold, a student-driven community formed on Twitter with the handle @BlackInBiophys and a logo designed by Taneisha Gillyard, a former postdoc in the Davis lab. Davis spoke at a virtual meeting held by the group.

A former postdoc in the Davis lab, Taneisha Gillyard, designed the logo for @BlackInBiophys, a student-driven community that formed on Twitter during the pandemic. Credit: Taneisha Gillyard.
In a short time, a strong sense of community developed among people who may have never met in person, but know a lot more about each other through social media, Davis says. People share grant writing tips, training and job opportunities, and generally celebrate the scientists, their contributions, and career options for the next generation.
The visibility may help change other statistics about Black researchers receiving less NIH funding and being cited less often than their white colleagues, Davis says.
Davis also teamed up with Meharry colleague Jennifer Cunningham-Erves to develop a funded community outreach project to address community concerns about vaccines. He has spoken about the basic science of mRNA at townhall-style community meetings, in person and virtual. The online recordings have reached people from Chicago to New York to Haiti.
The project collaborates with a consortium of more than 90 churches in middle Tennessee and Better Options TN, a community nonprofit organization. To understand concerns, the Meharry team interviewed people in the Southern United States. They developed and organized content on a frequently updated web site, https://yourcovidvaxfacts.com/en.
"We asked about their thoughts about the vaccine and the virus," Davis says. "The biggest one, particularly for Black Americans, was the distrust with government and healthcare. But I was very impressed with some of the questions that the public had. They weren't getting answers, and they wanted answers. If you remember, one of the major issues with people not getting a vaccine was that they thought it would affect their DNA. They just weren't familiar with mRNA."
Davis felt their concerns and trust issues as well. He initially was cautious about being vaccinated himself, waiting to see more data about its safety in people. "Even being a scientist, I was hesitant," he says. "I didn't want to be one of the first," he says. But as he explained the science and helped alleviate concerns of others, he also convinced himself to get the vaccine too.
Meanwhile, back in the lab after the pandemic disruptions, Davis and his team are working to improve health outcomes for populations most at risk, one variant protein and pathway at a time.
- Carol Cruzon Morton
0 notes
Text
Using MicroED for molecule conformations
This publication highlight is part of the SBGrid/Meharry Medical College Communities Project, focused on science education and demonstrating how structural biology and preclinical science connect to medicine.
Microcrystal electron diffraction (MicroED) is an emerging technique that has been shown to determine the solid-state structure of small molecules and small proteins in microcrystals. The ability to gain structural insight from microcrystals allows for a lower burden on crystallographers and opens a new range of target molecules that were previously unavailable due to their inability to form high-quality crystals. One such molecule is the commonly prescribed drug Mirabegron. Mirabegron is used to treat overactive bladder symptoms, yet a high quality structure of the molecule has eluded scientists due to its powdery crystalline state. SBGrid member Dr. Tamir Gonen, from UCLA, was able to determine a structure of Mirabegron using MicroED and sample a stable conformer state of this important drug.

Pictured above is Dog beta3 adrenergic receptor (grey) and Mirabegron (cyan). Mirabegron is shown deeply embedded in the binding site of this adrenergic receptor. PDB:7DH5. CC BY SBGRID
In a study published last month in Advanced Science, Prof. Gonen was able to show that Mirabegron has two distinct conformer states, a cis and trans, in a low energy crystal. This lattice unit is held together using a myriad of interactions between Mirabegron molecules, but a majority of the attraction is due to the demand to bury hydrophobic regions. While this arrangement is low energy and highly stable, it does not appear to be the conformer state found upon binding to target sites in proteins. The image above shows that Mirabegron buries deep into binding sites on proteins, and to do this it would need to go through a large conformational change from the states sampled in MicroED. These insights help to push forward our knowledge of the Mirabegron mechanism of action and could help describe the off-target effects of this drug.
- Vida Storm Robertson, Fisk University
Vida Storm Robertson is a Masters Student in Chemistry at Fisk University working in both solid-state and solution based structural determination techniques. He plans on starting a PhD program in biophysics next fall.
0 notes
Text
A look into specific interactions between Bruton’s Tyrosine Kinase (BTK) and its inhibitors
Note: This publication highlight is part of the SBGrid/Meharry Medical College Communities Project, focused on science education and demonstrating how structural biology and preclinical science connect to medicine.
Non-receptor tyrosine kinases (nRTK) are a subgroup of tyrosine kinases that are responsible for the phosphorylation of proteins. As the name suggests, tyrosine kinases work by transferring a phosphate group from ATP to the tyrosine residue of a protein. Non-receptor refers to the group of tyrosine kinases found within the cytosol of the cell, unlike receptor kinases, which are embedded into the cellular membrane. nRTKs are involved in many cell functions such as regulating cell growth and proliferation. They also play critical roles in immune system regulation. One specific nRTK that is involved in propagating signals from B cell receptors is known as Bruton’s tyrosine kinase (BTK). BTK’s importance was discovered when it was revealed that mutations in the Btk gene, the gene that encodes BTK, leads to the development of X-linked agammaglobulinemia (XLA), an immunodeficiency disease. BTK also plays essential roles in many diseases such as mantle cell lymphoma, chronic lymphocytic leukemia, Waldenstrom macroglobulinemia, small lymphocytic lymphoma, marginal zone lymphoma, and chronic graft-versus-host disease, just to name a few. These factors combined have made BTK inhibition a target of several drug therapies aimed at treating B cell malignancies. These therapies include the first in class BTK inhibitor, Ibrutinib, and the second-generation inhibitors Acalabrutinib, Zanubrutinib and Orelabrutinib. Although all of these therapeutics have seen success in clinical applications, specific interactions between the drugs and BTK are not well understood.

Above: Front and back views of BTK/Acalabrutinib complex (PDB: 8FD9) CC BY SBGRID.
In this work, SBGrid member Amy Andreotti and colleague David Lin reported the first structure of BTK in complex with Acalabrutinib. They also report a structure of BTK with Tirabrutinib, another second-gen BTK inhibitor that, at the time of this publication, is in clinical use in Japan and Taiwan but not yet FDA approved. When comparing their BTK/Acalabrutinib complex structure with a previously reported structure of BTK/Ibrutinib complex, the authors noted several regions where structural differences occur based on evaluation of RMSD values. Comparisons between BTK/Acalabrutinib and BTK/Tirabrutinib reveal large conformational differences in the activation loop that the authors attribute to the kinase undergoing dynamic fluctuations when bound to the drug, after further investigation of the previously reported structure of BTK/Tirabrutinib complex. Along with broad structure characterization, the authors also identified a few key residues that interact with the inhibitors. Combined, their work shows the need for further probing into how these drugs interact with BTK in order to fully examine how these inhibitors bind.
Read more in PLOS One
- KeAndreya Morrison, Meharry Medical College

1 note
·
View note
Text
A personal history of the great Crystallography pioneer Dr. Wayne Hendrickson
Note: This publication highlight is part of the SBGrid/Meharry Medical College Communities Project, focused on science education and demonstrating how structural biology and preclinical science connect to medicine.
SBGrid member Dr. Janet L. Smith writes a beautiful history of crystallography legend and pioneering physicist Wayne Hendrickson in the International Union of Crystallography Journal. In this commentary, Dr. Smith gives a wonderful history of Dr. Hendrickson’s academic work as he developed into the world class researcher we all know today. Hendricks recently won the Ewald Prize from the International Union of Crystallography and published a magnum opus of sorts accumulating his lifelong work on solving the phase problem titled “Facing the phase problem”. This article describes how Dr. Hendrickson and his colleagues, including Dr. Smith, approached the phase problem, created solutions for it, and applied those solutions to modern technologies that now solve this problem with general ease. Dr. Smith then provides vital personal chronologies to the discussions in her follow up article in which she describes how Hendrickson, over the course of his career, systematically attacked this problem and developed the methodologies for which he is now famous. This biographical retelling of his work helps to humanize this great scientist and visualize his impact on the field of structural biology. The combination of Dr. Hendrickson’s scientific anthology and Dr. Smith's historical retelling reads together as a great biographical story of how great scientists are born and evolve into the giants we know today. Dr. Hendrickson's monograph and Dr. Smith's commentary can both be found in the International Union of Crystallography Journal.
Pictured below is PDB entry 1HR3, the first structure Dr. Janet L. Smith and Dr. Wayne Hendrickson published together in 1983 at the Naval Research Laboratory.
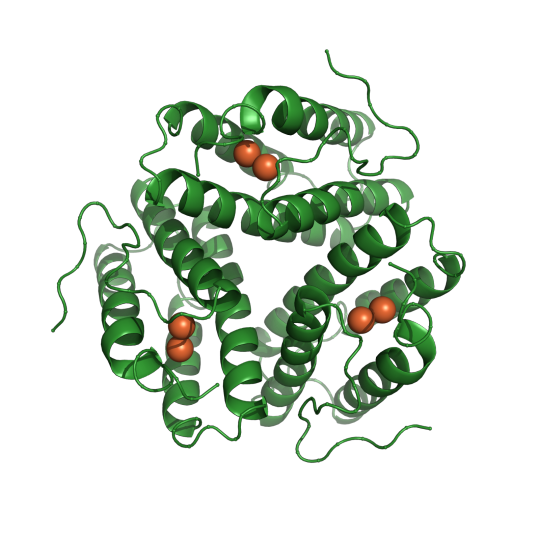
- Vida Storm Robertson, Fisk University
Vida Storm Robertson is a Masters Student in Chemistry at Fisk University working in both solid state and solution based structural determination techniques. He plans on starting a PhD program in biophysics next fall.
0 notes