#meiotic spindle
Explore tagged Tumblr posts
Visit Tumblr Blog
Explore Tumblr blogs with no restrictions, modern design and the best experience.
Last Seen Tumblr Blogs





Fun Fact
69% of Tumblr users are millennials.
Text

In a Spindle
Live imaging and electron tomography in cells of the tiny worm Caenorhabditis elegans (C.elegans for short) reveal fine details of the changes in cell structure occurring during female meiosis – specialised cell division for sexual reproduction to produce egg cells with only one pair of chromosomes. The length of the meiotic spindle (a transient assembly of proteins that forms to segregate chromosomes) is correlated with the length of microtubules – tiny flexible pipes formed of a protein called tubulin – which comprise most of the spindle proteins.
Read the published article here
Image from work by Vitaly Zimyanin and Stefanie Redemann
Department of Molecular Physiology and Biological Physics, University of Virginia School of Medicine, Charlottesville, VA, USA
Image originally published with a Creative Commons Attribution – NonCommercial – NoDerivs (CC BY-NC-ND 4.0)
You can also follow BPoD on Instagram, Twitter and Facebook
#science#biomedicine#immunofluorescence#biology#cells#meiosis#cytoskeleton#microtubules#meiotic spindle#c.elegans#nematodes
15 notes
·
View notes
Text
youtube
#R-loops#centromeres#meiosis#chromosome alignment#oocyte division#genome stability#chromosome segregation#epigenetics#molecular biology#RNA-DNA hybrid#genetic integrity#female fertility#oogenesis#reproductive biology#DNA regulation#gene expression#cell division#chromatin structure#aneuploidy#meiotic spindle.#Youtube
0 notes
Text
Propaganda!


Granulocytes are cells in the innate immune system characterized by the presence of specific granules in their cytoplasm. There are four types of granulocytes (full name polymorphonuclear granulocytes): Basophils, eosinophils, neutrophils, and mast cells. Except for the mast cells, their names are derived from their staining characteristics; for example, the most abundant granulocyte is the neutrophil granulocyte, which has neutrally staining cytoplasmic granules.
In cell biology, the spindle apparatus is the cytoskeletal structure of eukaryotic cells that forms during cell division to separate sister chromatids between daughter cells. It is referred to as the mitotic spindle during mitosis, a process that produces genetically identical daughter cells, or the meiotic spindle during meiosis, a process that produces gametes with half the number of chromosomes of the parent cell.
#Granulocytes#Spindle fibers#tournament poll#polls#wikipedia#cells of the human body#science tournament#biochemistry#poll#tumblr poll#tumblr polls
10 notes
·
View notes
Text
Decoding protein phosphorylation during oocyte meiotic divisions using phosphoproteomics
Oocyte meiotic divisions represent a critical process in sexual reproduction, as a diploid non-dividing oocyte is transformed into a haploid fertilizable egg, as a prelude for the subsequent embryonic divisions and differentiation. Although cell differentiation and proliferation are governed by transcription, oocyte maturation and early embryonic divisions depend entirely on changes in protein abundance and post-translational modifications. Here, we analyze the abundance and phosphorylation of proteins during Xenopus oocyte meiotic maturation. We reveal significant shifts in protein stability, related to spindle assembly, DNA replication and RNA-binding. Our analysis pinpoints broad changes in phosphorylation correlating with key cytological meiotic milestones, noteworthy changes in membrane trafficking, nuclear envelope disassembly and modifications in microtubule dynamics. Additionally, specific phosphorylation events target regulators of protein translation, Cdk1 and the Mos/MAPK pathway, thereby providing insight into the dynamics of Cdk1 activity, as related to the meiotic cell cycle. This study sheds light on the orchestration of protein dynamics and phosphorylation events during oocyte meiotic divisions, providing a rich resource for understanding the molecular pathways orchestrating meiotic progression in the frog, and most likely applicable to other vertebrate species. http://dlvr.it/TB7jLB
0 notes
Text
Mechanisms of Meiotic Spindle Initiation in Caenorhabditis elegans Oocytes
http://dlvr.it/T5nVWf
0 notes
Text
Unit 4b: How Meiosis Works
During meiosis, one cell divides twice, thus producing four cells, each with ½ the number of chromosomes. Therefore, each gamete has only ½ the genetic information. This makes the gametes haploid (as opposed to diploid).
Meoisis can be divided into 9 stages. The first time that the cell divides is meiosis I, and the second time it divides is meiosis II.
Background
A pair of homologous chromosomes is a set of one maternal and one paternal chromosome. These two chromosomes pair up with each other inside the cell during fertilization.
The two homologous chromosomes have the same:
gene sequence
gene loci
chromosomal length
centromere location
(But they differ in alleles.)
Each maternal chromosome has a corresponding paternal chromosome, and they pair up during meiosis.
The sex chromosomes (the 23rd pair) are homologous in females (XX) but not in males (XY).
Non-homologous chromosomes are called heterologous chromosomes.
Here is the first homologous pair of chromosomes (called Chromosome 1):
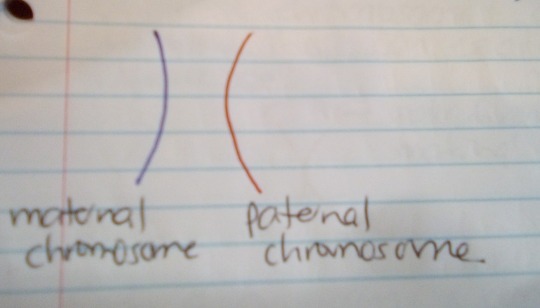
-
MEIOSIS I
Interphase
The DNA in the cell is copied (replicated). This results in 2 identical full sets of chromosomes.
Here are the original and duplicate of Chromosome 1 (1a is the original, and 1b is the duplicate):

There are two centrosomes outside of the nucleus, each containing a pair of centrioles. The centrosomes are essential for the process of cell division.
During interphase, microtubules extend from these centrosomes.
Prophase I
The copied chromosomes condense into X-shaped structures that can easily be seen under a microscope. The maternal chromosome pairs up with its duplicate, and the paternal chromosome pairs up with its duplicate.

Each chromosome is composed of 2 sister chromatids with identical genetic information.
The chromosomes now pair up so that the maternal pair & paternal pair from Chromosome 1 are together, etc.
The pairs of chromosomes may then exchange bits of DNA in a process called recombination or “crossing over”.
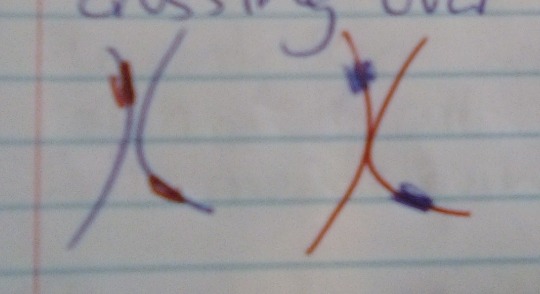
So now the 2 maternal chromosomes (in blue) are no longer completely identical, and neither are the 2 paternal chromosomes (in red).
At the end of this phase, the membrane around the cell nucleus dissolves away. This releases the chromosomes.
The meiotic spindle consists of microtubules and other proteins. It extends across the cell between the centrioles.
Metaphase I
The chromosome pairs line up next to each other along the equator (centre) of the cell.
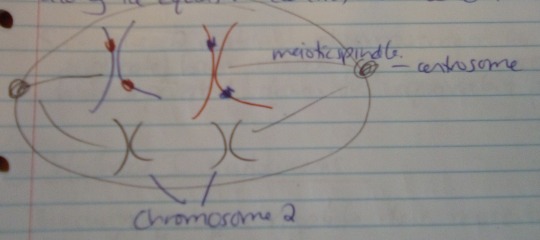
The centrioles are now at opposite poles of the cell, with the meiotic spindles extending from them.
The fibres of the meiotic spindle attach to one chromosome of each pair.
Anaphase I
The meiotic spindle then pulls the chromosome pairs apart, so that the maternal pair is pulled to one pole of the cell, and the paternal pair is pulled to the opposite pole.

The sister chromatids (eg the maternal chromosome & its duplicate) stay together in meiosis I (but this is not the case later in meiosis II).
In short – the 2 maternal chromosomes (original & duplicate) have paired up, and have been separated from the equivalent paternal pair.
Telephase I & cytokinesis
The chromosomes complete their move to opposite poles of the cell.
A full set of chromosomes gathers together at each pole. BUT one set is made up of the original maternal chromosomes & their duplicates (with a bit of paternal DNA due to the crossing-over), and vice versa for the other set.
The single cell pinches in the middle to form two separate daughter cells. Each one has a full set of chromosomes within a nucleus. This process is called cytokinesis.
-
MEIOSIS II
Prophase I
Now there are 2 daughter cells, each with a full set of 23 pairs of chromatids.
One cell has the maternal pairs (with some paternal DNA), and the other has the paternal pairs (with some maternal DNA).
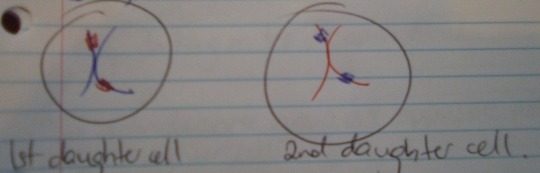
In each daughter cell, the chromosomes condense again into visible X-shaped structures (as seen above).
The membrane around the nucleus of each cell dissolves away. This releases the chromosomes, BUT they are still in their cells.
The centrioles duplicate, and the meiotic spindle forms again.
Metaphase II
In each cell, the chromosomes (i.e. pairs of sister chromatids) line up end-to-end along the cell equator).

In each cell, the centrioles are now at opposite poles.
Meiotic spindle fibres at each pole of the cell attach to each of the sister chromatids.

Anaphase II
The meiotic spindle pulls the sister chromatids APART, to opposite poles of the cell. In other words, the maternal pair are finally separated, and in the other cell, so are the paternal pair.
Now each separated chromatid is counted as an individual chromosome.
These separated chromatids complete their move to opposite poles of the cell. A full set of 23 individual chromosomes gathers at each pole.
A membrane forms around each set of chromosomes, which creates two new cell nuclei.
This is the last phase of meoisis, but another round of cytokinesis is still required.
Each daughter cell divides into two grand-daughter cells via cytokinesis. Now there are four grand-daughter cells from one original cell. Each is haploid, with 23 individual chromosomes.
In males, these four cells are all sperm cells.
In females, only one of these cells is an egg cell. The others are called polar bodies – small cells that don't develop into eggs.
-
Source: Science: NCEA Level 1 (New Zealand Pathfinder series).
4 notes
·
View notes
Text
types of chromosomal rearrangements and their consequences
while individuals differ by point mutations and small DNA sequence alterations, species differ from one another by chromosomal rearrangements (CRs)
types of CRs in the DNA sequence
bp-number change: large-scale deletions and duplications in chromosomal DNA sequence
less large-scale insertions because too difficult to synthesize independently without subsequent repair
change in segment’s location: inversions and non-homolog reciprocal translocations (i.e., large-portion recombination with non-homolog, completely unrelated chromosome) in chromosomal DNA
a CR type can be both a cause and an effect of another CR type
CR source - double-strand breakage in DNA sequence
double-strand breakage could be due to irradiation, mechanistic damage, or mutagen presence in cell/organism
1 chromosome → breakage → fragment undergoes no positional change → NHEJ after some end portions have degraded → deletion
1 chromosome → breakage → fragment flips in nuclear space → NHEJ → inversion
2 sister chromatids or homologs → breakage in both, at different locations → two fragments switch places in nuclear space → reparative recombination → sis_A + frag_B → sis_B + frag_A → sis_A deletion, sis_B duplication or vice versa
2 unrelated non-homologs → breakage in both → two fragments switch places in nuclear space → reparative recombination → NH_A + frag_B → NH_B + frag_A → reciprocal translocation of genes in frag_A and frag_B to different chromosomes entirely
CR source - aberrant crossover events between repeats along DNA sequence
repeated segments - simple sequence repeats (SSRs) that usually direct recombination enzymes OR duplicate, mutable transposable elements
**1 chromosome → 2 repeats with the same polarity, DNA region in between → recombination with self → segment in between repeats is looped out → deletion
**1 chromosome → 2 repeats with opposing polarities, DNA region in between → recombination with self → segment in between repeats is joined to different ends → inversion
2 sister chromatids or homologs → 2 repeats, each at a different position → recombination → sis_A + frag_B → sis_B + frag_A → sis_A deletion, sis_B duplication or vice versa
2 unrelated non-homologs → 2 repeats → recombination → NH_A + frag_B → NH_B + frag_A → reciprocal translocation of genes in frag_A and frag_B to different chromosomes entirely
“opposing polarities” of two identical repeats, in itself represents inverted repeat sequences
CR detection - experimental methods
detecting chromosomal rearrangement in organisms
in-situ fluorescent hybridization (FISH) - stains non-origin chromosomal DNA a different color than the rest of an altered chromosome in a method called “barcoding” for unique chromosomal identifiers along DNA sequence
gDNA microarray - small radio/fluorescent-labeled DNA segment is mixed with fragmented genomic library to analyze degree of hybridization to non-origin, altered chromosome
genome nucleotide sequencing - reveals exact rearrangement break points and DNA sequence alteration down to the base-pair level
PCR nucleotide sequencing - also reveals rearrangement break points, via analysis of specific DNA band presences after sequence amplification
research utility of deletion mutations
deletion mapping with known deletion sequences and resulting mutant phenotypes allows for experimental gene location along chromosome
gene occurring in between two deletion locations can be found using deletion mutant phenotypes as long as endpoint locations of deletions are known
research utility of inversion mutations
inversions block viable/successful recombination event occurrence (see peri- and paracentric inversion heterozygote outcomes below)
inversions can be used to generate “balancer chromosomes” and corresponding visibly dominant phenotypes in experiments
CR effects - gene and phenotype level
for most chromosomal rearrangement homozygotes, recombination is not a problem - F1 heterozygote recombination generates abnormalities
deletions: effects on DNA replication in heterozygote
in meiosis, the homolog’s corresponding DNA segment to the deleted portion never recombines - affects map distances
previously unfound deletions can be detected during meiosis, when homolog DNA forms “deletion loop”
all genes along loop cannot recombine with the deleted homologous segment and thus always segregate together
thus, look for consistent parental type gene segregation, which indicate that observed genes are within deletion loops
deletions: effects on heterozygote phenotype
hemizygous “heterozygote” may display dominant WT, recessive WT due to haploinsufficiency
hemizygous “heterozygote” is vulnerable to environmental DNA damage and subsequent rise of mutant phenotype during lifetime
already-existing recessive homolog mutation in hemizygous “heterozygote” may be uncovered after deletion of WT gene in chromosome
duplications: effects on DNA replication in heterozygote
chromosome with tandem, adjacent repeats in meiosis → one repeat is looped out in recombination → 1 of 2 duplicates deleted
chromosome with tandem repeats in meiosis → repeats recombine along same chromosome, with each other → both repeats are removed → homolog’s repeat segment is looped out during meiosis, like a deletion loop
chromosome with non-tandem, dispersed repeats in meiosis → aberrant recombination event-like occurrence in meiotic recombination**
unequal crossing over creates crossover = 2 repeats • 2 repeats → 1 repeat and 3 repeats
duplications: effects on heterozygote phenotype
mostly non-expressed in phenotype due to reconcilable nature of gene dosage
mutant phenotype expressed for protein functions particularly sensitive to gene dosage levels
new physical chromosomal location may alter gene function because DNA’s external environment can play a role in regulating gene expression
extremely large duplications have serious phenotypic consequences
inversions: effects on DNA replication in heterozygote
recombination between WT and inverted area - inversion loop forms between inverted chromosome segment and WT homologous corresponding segment, and crossover point occurs within the loop
results in formation of genetically unequal F2 chromosomes, i.e., reduced fertility of F1 heterozygote
for pericentric inversions, in which centromere falls within inversion loop - one recombinant chromatid has a deleted portion | the other has a duplicated portion
for paracentric inversions, in which centromere is outside inversion loop - one recombinant chromatid loses a centromere and is acentric, degrading as a non-chromosome fragment | the other is dicentric, chromosomal but pulled into fragments by opposing spindle fibers during metaphase I
affects map distances - inversion loop recombination is never successful and so distances calculated from RF between inverted genes always = 0 cM
inversions: effects on heterozygote phenotype
mostly non-expressed because segments still encode genes properly
unless! inversion break points fall in the middle of a gene, which results in dys- or non-functional gene due to separation of two previously-connected portions
reduction in inversion heterozygote fertility varies directly with size of inversion (crossing over produces nonviable recombinant gametes, and larger DNA segment size = larger probability of crossover occurrence)
reciprocal translocation: effects on DNA replication in heterozygote
P generation segregates normally into translocation parent (_____-------) + (--------______) haploid gamete x WT parent (__________) + (--------------)
F1 diploid zygote has genotype (_____-------) + (--------______) and (__________) + (--------------), which cannot independently pair up in meiosis I because of translocated segments
translocated chromosomes (T1, T2) and normal chromosomes from other parent (N1, N2) form cruciform meiotic pairing structure to recombine for metaphase I
reciprocal translocation: effects on DNA replication and phenotype in heterozygote
post-cruciform segregation results in possible formation of genetically unequal F1 gametes - semisterility of F1 heterozygote, depending on occurrence of:
alternate segregation pattern: chromosomes diagonal from one another segregate together in meiosis I to form balanced, viable gametes (occurs in ~50% of translocation heterozygotes)
adjacent segregation patterns 1 & 2: chromosomes segregate with either one of adjacent chromosomes in meiosis I to form inviable, unbalanced gametes
pseudolinkage occurs in genes at translocation break points due to P(segregation patterning) alterations in RFs and mapping
if ---(a)---(b)--- and ___(c)___(d)___ undergo reciprocal translocation and create ---(a)-__(d)___ and ___(c)_--(b)---, the heterozygote with a+d / c+b translocation genotype generates
gamete with both translocate chromosomes: a+d / c+b
gamete with both WT chromosomes: a+b / c+d
gamete with 1 translocate and 1 WT chromosome: a+d / c+d no b chromosomal arm, so inviable
gamete with 1 translocate and 1 WT chromosome: a+b / c+b no d chromosomal arm, so inviable
13 notes
·
View notes
Text
Coenzyme Q10 improves the in vitro maturation of oocytes exposed to the intrafollicular environment of patients on fertility treatment.
PMID: JBRA Assist Reprod. 2020 Mar 23. Epub 2020 Mar 23. PMID: 32202744 Abstract Title: Coenzyme Q10 improves the in vitro maturation of oocytes exposed to the intrafollicular environment of patients on fertility treatment. Abstract: OBJECTIVE: To evaluate the impact of patient follicular environment with oxidative stress on oocyte quality.METHODS: Patients on fertility treatment with either advanced maternal age or endometriosis were asked to donate follicular fluid collected during ovum pick-up. Follicular fluid (FF) was added (20%, 10% and 5%; %V/V) to in vitro maturation (IVM) medium with mouse oocytes. Following maturation culture, the oocytes were assessed for meiosis reinitiation. In a second setup, coenzyme Q10 was added to culture medium with FF. In addition to assessing meiotic maturation, a subset of oocytes was assessed for spindle structure and chromosome alignment.RESULTS: Supplementation of IVM medium with FF of patients of advanced maternal age (with or without antioxidants) did not have an effect on the maturation capacity of mouse oocytes. However, the addition of FF of individuals with endometriosis (without antioxidants) in the two highest concentrations affected oocyte maturation (61.5%&57.0% maturation) compared with the lowest concentration (89.2% maturation) (p
read more
0 notes
Link
Chromosomes become visible, crossing-over occurs, the nucleolus disappears, the meiotic spindle forms, and the nuclear envelope disappears. To see prophase I animated, click the Play button. ... The duplicated homologous chromosomes pair, and crossing-over (the physical exchange of chromosome parts) via Lakhasly.com Rss Feed
0 notes
Text
Male meiotic spindle features that efficiently segregate paired and lagging chromosomes
http://dlvr.it/RBVKTt
0 notes
Text
Importin α and vNEBD Control Meiotic Spindle Disassembly in Fission Yeast.
Pubmed: http://dlvr.it/QQrd5k
0 notes
Text
Meiotic spindle formation in mammalian oocytes: Implications for human infertility.
Pubmed: http://dlvr.it/QBkXDB
0 notes
Text
Zap70 and downstream RanBP2 are required for the exact timing of the meiotic cell cycle in oocytes.
Zap70 and downstream RanBP2 are required for the exact timing of the meiotic cell cycle in oocytes. Cell Cycle. 2017 Jul 26;:0 Authors: Kim HJ, Lee SY, Lee HS, Kim EY, Ko JJ, Lee KA Abstract In previous studies, we observed that Zeta-chain-associated protein kinase 70 (Zap70) regulates spindle assembly and chromosome alignment in mouse oocyte and that Ran binding protein 2 (RanBP2) is highly associated gene with Zap70 based on a microarray analysis. Because RanBP2 is related to nuclear envelope breakdown (NEBD) during mitosis, the aim of the present study was to elucidate the molecular mechanism of Zap70 with respect to RanBP2 in the germinal vesicle breakdown (GVBD) of oocytes. Results indicated that RanBP2 expression was regulated by Zap70 and that depletion of RanBP2 using RanBP2 RNAi manifested comparable phenotypes to those observed in Zap70 RNAi-treated oocytes, which presented faster processing of GVBD. Additionally, Zap70 RNAi-treated oocytes showed faster meiotic resumption with premature activation of maturation-promoting factor (MPF), premature division of chromosomes at approximately 6-8 h and more rapid degradation of securin. In conclusion, we report that Zap70 is a crucial factor for controlling the exact timing of meiotic progression in mouse oocytes. PMID: 28745977 [PubMed - as supplied by publisher] http://dlvr.it/PYtJ7R
0 notes
Text
The effect of B chromosomes on meiotic and pre-meiotic spindles and chromosome pairing in Triticum/Aegilops hy
http://dlvr.it/P7w0D8
0 notes
Text
Vitamin C protects carboplatin-exposed oocytes from meiotic failure.
PMID: Mol Hum Reprod. 2019 Aug 8. Epub 2019 Aug 8. PMID: 31393565 Abstract Title: Vitamin C protects carboplatin-exposed oocytes from meiotic failure. Abstract: CBP (carboplatin) is a second-generation chemotherapeutic drug of platinum compound commonly applied in the treatment of sarcomas and germ cell tumours. Although it is developed to replace cisplatin, which has been proven to have a variety of side effects during cancer treatment, CBP still exhibits a certain degree of toxicity including neurotoxicity, nephrotoxicity, hematotoxicity and myelosuppression. However, the underlying mechanisms regarding how CBP influences the female reproductive system especially oocyte quality have not yet been fully determined. Here, we report that CBP exposure led to the oocyte meiotic defects by impairing the dynamics of the meiotic apparatus, leading to a remarkably aberrant spindle organisation, actin polymerisation and mitochondrial integrity. Additionally, CBP exposure caused compromised sperm binding and fertilisation potential of oocytes by due to an abnormal distribution of cortical granules and its component ovastacin. More importantly, we demonstrated that vitamin C supplementation prevented meiotic failure induced by CBP exposure and inhibited the increase in ROS levels, DNA damage accumulation and apoptotic incidence. Taken together, our findings demonstrate the toxic effects of CBP exposure on oocyte development, and provide a potential effective way to improve the quality of CBP-exposed oocytes in vitro.
read more
0 notes
Text
Tea polyphenol protects against cisplatin-induced meiotic defects in porcine oocytes.
PMID: Aging (Albany NY). 2019 Jul 13 ;11(13):4706-4719. PMID: 31301169 Abstract Title: Tea polyphenol protects against cisplatin-induced meiotic defects in porcine oocytes. Abstract: DDP (cisplatin), a DNA cross-linking agent, is one of the most common chemotherapeutic drugs that have been widely used in the treatment of sarcomas and germ cell tumors. DDP treatment exhibits severe side effects including renal toxicity, ototoxicity and embryo-toxicity. Women of reproductive age treated with DDP may lead to loss of primordial follicles, resulting in the depletion of the ovarian reserve and consequent premature ovarian failure. However, the influence of DDP on the oocyte quality and the strategy to prevent it has not yet fully clarified. Here, we report that DDP exposure resulted in the oocyte meiotic failure via disrupting the meiotic organelle dynamics and arrangement, exhibiting a prominently impaired cytoskeleton assembly, including spindle formation and actin polymerization. In addition, exposure to DDP led to the abnormal distribution of mitochondrion and cortical granules, two indicators of cytoplasmic maturation of oocytes. Conversely, TP (tea polyphenols) supplementation partially restored all of the meiotic defects resulted from DDP exposure through suppressing the increase of ROS level and the occurrence of DNA damage as well as apoptosis.
read more
0 notes