#eu mdr ce marking certification for medical devices
Explore tagged Tumblr posts
Visit Tumblr Blog
Explore Tumblr blogs with no restrictions, modern design and the best experience.
Last Seen Tumblr Blogs





Fun Fact
Celebrities use Tumblr as well.
Text
Key Regulations Followed By Laparoscopic Instruments Manufacturer In India
Laparoscopic instrument manufacturers must adhere to stringent regulations to ensure the safety, effectiveness, and quality of their products. These regulations are enforced by various national and international health authorities to ensure that laparoscopic devices meet medical standards for patient safety. Here are some of the key regulations followed by Laparoscopic Instruments Manufacturer In India:

1. ISO Standards for Quality Management
ISO 13485 Certification: This is the internationally recognized standard for quality management systems specific to medical device manufacturers. It ensures that laparoscopic instruments are designed, developed, and manufactured under stringent quality controls.
ISO 14971 – Risk Management: Manufacturers must comply with this standard to manage the risks associated with the design and production of laparoscopic instruments. This includes risk identification, analysis, and mitigation throughout the product lifecycle.
2. FDA (Food and Drug Administration) Regulations
FDA 21 CFR Part 820 – Quality System Regulation (QSR): Manufacturers selling in the U.S. must comply with the FDA’s QSR, which outlines requirements for designing, manufacturing, packaging, and distributing medical devices.
Premarket Approval (PMA): For certain Class III medical devices, manufacturers must obtain FDA approval through the PMA process, demonstrating the safety and effectiveness of the laparoscopic instruments before they can be marketed.
510(k) Clearance: Many laparoscopic instruments fall under Class II devices, requiring manufacturers to submit a 510(k) premarket notification to the FDA. This process requires proving that the new device is substantially equivalent to an already legally marketed device.
3. CE Marking and European Union Medical Device Regulations (MDR)
CE Mark Certification: For manufacturers selling in Europe, obtaining the CE mark is mandatory.
EU MDR (Medical Device Regulation): The EU MDR requires manufacturers to provide comprehensive documentation, including clinical evidence of the device’s safety and performance. It also emphasizes post-market surveillance and reporting of adverse events.
4. Good Manufacturing Practices (GMP)
Compliance with GMP Guidelines: Manufacturers must follow GMP practices, which ensure that laparoscopic instruments are consistently produced and controlled according to quality standards. This includes maintaining cleanroom environments, proper equipment maintenance, and employee training.
Traceability and Documentation: GMP also requires maintaining detailed records of production batches, testing, and quality checks to ensure traceability and accountability.
Rudra Surgical is known as Laparoscopic Simulator Virtual Endo Trainer Manufacturer in India. If you or anyone you might know is searching for a reliable manufacturer of laparoscopic instruments, then you can connect with Rudra Surgical.
About Rudra Surgical
Rudra Surgical is one of the leading names because it offers a wide range of medical equipment by designing it precisely. To know more details about various medical equipment, you can connect with them without any delay.
#Laparoscopic Instruments Manufacturer In India#Laparoscopic Simulator Virtual Endo Trainer Manufacturer in India
0 notes
Text
What is CE Mark Certification, and why is it important for products sold in Ireland?
/ Uncategorized / By deepika

Understanding CE Mark Certification in Ireland
CE Mark Certification in Ireland Ensuring that products meet protection, fitness, and environmental standards is essential in the global marketplace. For merchandise provided inside the European Economic Area (EEA), in conjunction with Ireland, CE Mark Certification in Ireland serves as a critical compliance indicator. This blog explores what CE Mark Certification Certification in Ireland is, why it’s crucial, and how it impacts merchandise supplied in Ireland.
What is CE Mark Certification?
CE Mark Certification in Ireland is a necessary conformity mark for favourable products offered inside the EEA. The “CE” stands for “Conformité Européenne,” which is interpreted as “European Conformity.” This certification indicates that a product complies with necessary European fitness, safety, and environmental requirements, permitting it to be marketed in all EEA nations and Ireland.
The CE mark Certification in Ireland isn’t a notable guarantee mark, but rather an assertion by the manufacturer that their product meets the EU’s regulatory necessities. It is a visible image that assures clients and regulatory authorities that the product adheres to EU regulations.
The Purpose of CE Mark Certification in Ireland
The number one motive of CE Mark Certification in Ireland is to ensure an immoderate degree of safety and protection for customers, in addition to facilitating the loose motion of products in the EEA. Here’s a better examine why CE Certification is crucial:
Consumer Safety: The CE mark Certification in Ireland guarantees that products meet strict EU requirements related to fitness and protection. For example, electric domestic gadgets need to meet necessities to save you from electric shocks or fires; at the same time, toys need to be examined for choking dangers. This protects clients from risky products and promotes bearing in mind the goods they purchase.
Legal Requirements: CE marking is a prison requirement for many product instructions to be promoted inside the EEA. Without the CE mark Certification in Ireland , merchandise cannot be legally advertised or offered, which may cause fines, product remembers, or even a ban on selling the product in the market.
Market Access: The CE mark gives the European marketplace the right to enter one of the world’s most critical and rewarding markets. For manufacturers and importers, it simplifies the method of getting into new markets within the EEA, decreasing the need for more than one certification in exceptional nations.
Product Confidence: The CE mark Certification in Ireland is a stamp of super and compliance. It shows clients and industrial organization companions that the product has been evaluated and meets rigorous necessities. This can decorate a product’s reputation and foster patron self-guarantee.
The CE Mark Certification Process in Ireland
Obtaining CE Mark Certification in Ireland entails several vital steps, which producers and importers need to conform with:
Identify Applicable Directives and Regulations: Different products are subject to unique EU directives and tips. Manufacturers need to discover which particular directives they shook their products. For instance, medical devices fall under the Medical Devices Regulation (MDR), while the Machinery Directive rules machines.
Assess Conformity: Once the applicable directives are recognized, producers must check their product’s conformity to the necessities. This regularly entails conducting exams and reviews to ensure the product meets EU requirements.
Compile Technical Documentation: Manufacturers want to prepare and hold a technical record that includes format and manufacturing facts, threat tests, and check evaluations. This report demonstrates how the product meets EU requirements.
Declaration of Conformity: The manufacturer should draft and sign an EU Declaration of Conformity, a file that broadcasts the product’s compliance with relevant directives and guidelines.
Affix the CE Mark: Once all necessities are met, the CE mark Certification in Ireland may be affixed to the product. This entails placing the mark on the product itself or its packaging in a visible, legible, and indelible way.
Involve a Notified Body (if required): For high-quality merchandise, especially humans with a higher chance, a Notified Body (an independent agency precise by EU global locations) is needed. The Notified Body performs reviews and audits to ensure compliance in advance rather than issuing certification.
Impact of CE Mark Certification on Products Sold in Ireland
For products offered in Ireland, CE Mark Certification in Ireland has numerous sizable impacts:
Regulatory Compliance: The CE mark Certification guarantees that products meet EU tips, which is critical for criminal income in Ireland. Non-compliance can motivate criminal outcomes and preclude market access.
Market Confidence: Products with the CE mark Certification in Ireland are considered safer and more dependable. This may affect patron preference and beautify the product’s competitiveness in the Irish marketplace.
Trade Facilitation: For corporations exporting to Ireland, CE Mark Certification in Ireland simplifies the alternate approach by harmonizing standards throughout the EU. It reduces the need for introduced certifications or checking out, particularly in Ireland, streamlining market access.
Liability and Risk Management: Complying with CE necessities permits producers and importers to control criminal duty risks by ensuring that merchandise meets safety requirements. This can mitigate the danger of product recollects, legal claims, and damage to logo reputation.
Conclusion
CE Mark Certification in Ireland is vital in ensuring that merchandise meets important protection, health, and environmental requirements earlier than accomplishing the market. For products offered in Ireland, acquiring CE Certification guarantees compliance with EU rules and complements the marketplace admission to customers who endure in thought and not unusual product safety. Understanding and adhering to the CE marking approach is vital for organizations aiming to efficiently navigate the European market and defend their products and popularity.
For agencies looking to enter or expand into the Irish market, investing time and resources into obtaining CE Certification is a strategic step that may result in stepped-forward market possibilities and long-term fulfillment.
Why Factocert for CE Mark Certification in Ireland
We provide the best CE Mark consultants in Ireland, who are very knowledgeable and provide the best solutions. To know how to get ISO certification in Ireland, kindly reach us at [email protected]. ISO Certification consultants work according to ISO standards and help organizations implement CE Mark Auditors in Ireland with proper documentation.
For More Information Visit, CE Mark Certification in Ireland
Related Links
ISO Certification in Ireland
ISO 9001 Certification in Ireland
ISO 14001 Certification in Ireland
ISO 45001 Certification in Ireland
ISO13485 Certification in Ireland
ISO 27001 Certification in Ireland
ISO 22000 Certification in Ireland
CE Mark Certification in Ireland
Halal certification in Ireland
0 notes
Text
What is CE MARK Certification in Bloemfontein?

What is CE MARK Certification in Bloemfontein?
CE MARK Certification in Bloemfontein stamp is vital for Bloemfontein businesses aiming for European trade. “Conformité Européenne,” or CE MARK, signifies a product’s conformity with the European Union’s (EU) rigorous safety, health, and eco-friendly standards. For Bloemfontein maker-exporters, getting a CE MARK certificate is pivotal for entry into the prestigious European Economic Area (EEA) – this zone covers all EU countries and many more worldwide.
CE MARK certification in Bloemfontein is a regulatory requirement for pleasant and safe diagnoses globally. It assures customers and regulators that the product complies with European requirements, making it a critical step for any organization seeking to compete worldwide.
Understanding CE MARK Certification
The CE MARK Certification in Bloemfontein is obligatory for a massive style of products, which incorporates electronics, devices, medical gadgets, and manufacturing products. It is a way for a product to be assessed and meet the crucial requirements of applicable EU directives. The technique includes rigorous trying out, documentation, and auditing to ensure the product is stable for use and meets the vital requirements.
In CE MARK Certification in Bloemfontein, corporations trying to acquire CE MARK certification in Bloemfontein want to apprehend the precise directives that are observed in their products. This approach may be complex, as each product’s magnificence may have first-rate necessities. For instance, scientific gadgets are ruled through the Medical Device Regulation (MDR), while virtual gadgets must comply with the Electromagnetic Compatibility (EMC) Directive.
CE MARK Consultants in Bloemfontein
Navigating the CE MARK certification in Bloemfontein method may be challenging, especially for agencies new to the European marketplace. This is where CE MARK Certification in Bloemfontein play a critical role. These specialists are interested in helping corporations understand and observe the EU’s complicated regulatory framework.
Services Provided through CE MARK Consultants in Bloemfontein:
1. Regulatory Guidance: CE MARK Certification in Bloemfontein professionals in Bloemfontein offer professional advice on the specific EU directives that you have a look at in your products. They help corporations understand the crook necessities and ensure that their merchandise meets all critical necessities.
2. Product Testing and Validation: Consultants coordinate with joint laboratories to carry out the favoured checks on products. This includes safety testing, electromagnetic compatibility testing, and unique checks necessary for CE MARK certification.
3. Technical Documentation: Preparing the technical report is essential to the CE MARK certification in Bloemfontein technique. This document must encompass all relevant product specifications, threat tests, and test results. CE MARK experts assist in compiling this documentation to ensure it meets EU requirements.
4. Conformity Assessment: Consultants guide agencies via the conformity evaluation method, comprising self-evaluation or zero.33-birthday celebration certification, depending on the product class.
5. Training and Education: CE MARK specialists often offer schooling for employer personnel, helping them apprehend the certification method, regulatory necessities, and the way to hold compliance.
6. Audit Preparation: Before the final audit, experts conduct internal audits to identify functionality issues. This ensures that corporations are prepared for the actual CE MARK audit.
CE MARK Auditors in Bloemfontein
CE MARK auditors in Bloemfontein are chargeable for conducting thorough opinions of merchandise and their related documentation to ensure compliance with EU standards. The audit gadget is a vital step in obtaining CE MARK certification in Bloemfontein.
The CE MARK Audit Process:
1. Documentation Review: Auditors compare the technical report and relevant documentation to ensure completeness and accuracy. This consists of checking the product’s format specifications, hazard exams, and proof of compliance with applicable requirements.
2. On-Site Inspections: Auditors may moreover conduct on-net page inspections to affirm that the manufacturing approach aligns with documented strategies and that the product is constantly produced consistent with EU necessities.
3. Product Testing Validation: If the product trying out has been completed, auditors validate the effects to ensure compliance with EU requirements.
4. Audit Reporting: The auditor offers an intensive record outlining their findings after the audit. The auditor will propose issuing the CE MARK certification in Bloemfontein if the product meets all necessities.
5. Certification Issuance: The CE MARK certification in Bloemfontein is issued upon a triumphant crowning glory of the audit, permitting the product to be legally supplied within the EEA.
Why Factocert is the Best Provider of CE MARK Certification in Bloemfontein
Factocert is a prime agency of CE MARK certification services in Bloemfontein. Here’s why Factocert can be an excellent desire for companies on the lookout for CE MARK certification:
1. Expertise and Experience: Factocert has a group of pretty expert and skilled CE MARK professionals and auditors who are well-versed in the intricacies of EU guidelines. Their in-depth information ensures that corporations get a preserve of accurate steerage and help at some point in the certification way.
2. Comprehensive Services: Factocert offers a complete range of offerings, from regulatory guidance and product attempts to technical documentation schooling and audit helpful aid. Their forestall-to-end answers simplify the certification technique, making it less complicated for corporations to acquire compliance.
3. Customized Solutions: Factory is familiar with the fact that each employer is unique. They provide tailored answers that meet the precise desires of each customer, ensuring that their merchandise takes a look at applicable EU directives efficiently and effectively.
4. Proven Track Record: Factocert has a tested tune record of correctly assisting organizations in Bloemfontein to accumulate CE MARK certification. Their determination to excellence and client pleasure has earned them the acceptance of numerous corporations through several industries, CE MARK Certification in Bryanston.
5. Local Presence with Global Reach Factocert combines close understanding with worldwide achievement, making them sincerely exceptional accomplices for organizations in Bloemfontein searching for an increase in the European market. Their sturdy connections with global checking-out laboratories and certification bodies ensure a smooth certification approach.
Advantages of CE MARK Certification in Bloemfontein
Obtaining CE MARK certification gives several blessings for groups in Bloemfontein:
1. Market Access: CE MARK certification is a crook requirement for selling products within the European Economic Area (EEA). It opens up entry to over 30 countries, presenting groups with a large market for their merchandise.
2. Enhanced Consumer Confidence: The CE MARK is diagnosed globally as a symbol of safety and exquisite. Products with CE MARK certification are much more likely to be relied on through using consumers, which is necessary to extend profits and logo loyalty.
3. Competitive Advantage: CE MARK certification units a product apart from non-certified competition in a worldwide market. It demonstrates a determination to meet excessive protection and incredible requirements, giving businesses a competitive element.
4. Legal Compliance: CE MARK certification ensures that a product complies with EU pointers, lowering the danger of prison problems, fines, or product recalls. This compliance is critical for retaining popularity and avoiding highly-priced jail-annoy situations.
5. Streamlined Export Process: Once a product has CE MARK certification, it can be sold freely in the EEA without the want for additional checking out or certification. This simplifies the export process and reduces the obstacles to access within the European marketplace.
6. Increased Brand Value CE MARK certification enhances an emblem’s fee by associating it with high necessities of safety and best. This certification can result in more emblem recognition and credibility, each in Bloemfontein and internationally, CE MARK Certification in Bryanston.
7. Long-Term Business Growth: By obtaining CE MARK certification, corporations in Bloemfontein function themselves for sustainable growth. This certification opens doorways to new markets and income streams, fostering extended-time period success.
8. Encouragement of Innovation The machine attaining CE MARK certification regularly drives innovation in product format and manufacturing techniques. Businesses should adopt new technologies and practices that enhance product safety and standard overall performance, CE MARK Certification in Cape Town .
Conclusion
CE MARK certification isn’t the most effective felony requirement for organizations looking to enter the European marketplace; it’s also an effective device for demonstrating a determination to be safe. For companies in Bloemfontein, acquiring CE MARK certification with the help of expert professionals and auditors just like the ones at Factocert can launch new possibilities for growth and fulfilment.
Factocert’s expertise, whole services, and willpower to patron pleasure are why they best prefer CE MARK certification in Bloemfontein. By undertaking this certification, groups can extend their reap to the European marketplace with a chunk of successes, decorate their logo price, and increase for a certain prolonged period, CE MARK Certification in Pretoria .
Why Factocert for ISO Certification in Bloemfontein?
We provide the best ISO consultants Who are knowledgeable and provide the best solution. And to know how to get ISO certification. Kindly reach us at [email protected]. work according to ISO standards and help organizations implement ISO certification in India with proper documentation.
For More Information Please Visit CE MARK Certification in Bloemfontein.
Related Article: CE MARK Certification
0 notes
Text
What is CE Certification? Why is CE Mark Certification in Abu Dhabi Important?

CE Certification in Abu Dhabi:
CE Mark Certification in Abu Dhabi is key for businesses wanting to sell products in the European Economic Area (EEA) and more. It proves that a product meets important health, safety, and environmental protection standards in the European Union (EU). For Abu Dhabi firms, gaining CE certification can unlock European markets and build trust in areas that value the CE mark. This piece explores the relevance of CE certification, how to get it, and the positives it offers for Abu Dhabi businesses.
What is CE Certification?
The CE (Conformité Européenne) mark is significant. It means the maker says their product meets necessary European standards. You’ll find this mark on various items like toys, machines, and even scientific tools. This symbol shows the product meets the ideals and can be sold legally throughout the EEA.
Why is CE Certification in Abu Dhabi Important?
CE Mark certification in Abu Dhabi isn’t always a regulatory requirement for access to European markets; however, a mark of top-notch protection is identified globally. For organizations in Abu Dhabi, acquiring CE certification can beautify logo reputation, enhance product excellence, and grow market reach. It guarantees that products meet stringent European necessities, which are often benchmarks of excellence in extraordinary components of the arena.
The Process of CE Certification in Abu Dhabi
Step 1: Identify the Applicable Directive: The first step in the CE Mark certification in Abu Dhabi method is to become aware of which European directives or hints are observed in your product. Several directives are counting on product elegance, such as the Low Voltage Directive (LVD), Electromagnetic Compatibility (EMC) Directive, Medical Device Regulation (MDR), and others. Each directive outlines specific requirements that have to be met for compliance.
Step 2: Conduct a Conformity Assessment: The following step evaluates conformity once the relevant directive is diagnosed. This involves trying out the product to ensure it meets the necessities cited within the directive. Depending on the product class, this will embody electric protection assessments, mechanical assessments, chemical evaluations, and distinctive critiques. The assessment may be accomplished internally with the producer’s helpful resources or through a certified zero.33-birthday celebration attempting out body.
Step three: Compile Technical Documentation: Technical documentation is critical to the CE certification in Abu Dhabi approach. It must include precise facts about the product, format, and manufacturing records, danger checks, check evaluations, and customer manuals. This documentation proves that the product complies with the applicable directives and must be saved for at least ten years after the product is placed on the market.
Step 4: Affix the CE Mark: The CE mark certification in Abu Dhabi can be affixed to the product as quickly because the product has correctly passed the conformity evaluation and the technical documentation is complete. The CE mark certification in Abu Dhabi wants to be visible, legibly, and indelibly on the product, packaging, or accompanying documentation. It is a method that ensures that the product meets all requirements and may be legally supplied within the EEA.
Step 5: Issue a Declaration of Conformity: The last step within the CE Mark certification in Abu Dhabi technique is to problem a Declaration of Conformity (DoC). This is a correct declaration through the producer that the product complies with all relevant directives and has passed the crucial conformity assessments. The DoC needs to be signed with the beneficial, precious, practical, and helpful resource of an accountable person in the business organization agency and saved on the report when the technical documentation is issued.
Challenges in Obtaining CE Certification in Abu Dhabi
While the CE Mark Certification in Abu Dhabi approach is standardized at some point in the EU, Abu Dhabi businesses might also face specific annoying conditions because of network hints, market situations, and logistical issues.
Understanding EU Directives: One of the primary worrying conditions is understanding the complicated array of EU directives and how they practice unique merchandise. Each directive has its set of necessities, and agencies need to have radical information on these to ensure compliance. This may also require specialized facts or consultation with experts familiar with each EU and community guidelines.
Testing and Certification Costs: Another project is the charge associated with locating out and certification. Depending on the product’s elegance, the conformity assessment procedure may be luxurious, particularly if 1/3-party trying out is wanted. Those prices may be a widespread burden for small and medium-sized firms (SMEs) in Abu Dhabi. However, the funding is regularly justified through the potential market possibilities CE Certification in Abu Dhabi can free up.
Supply Chain Considerations: For businesses that depend on imported components or materials, ensuring that every additive of the product meets EU standards can take time. The entire supply chain should be scrutinized to ensure compliance, which may also require extra testing or verification from companies. This may be particularly complicated for products with several additives sourced from splendid global places.
The Role of Notified Bodies: In some instances, merchandise can also require the involvement of a Notified Body for the CE Mark certification in Abu Dhabi gadget. Notified Bodies are impartial organizations that are positive via EU member states to evaluate the conformity of excessive high-quality merchandise in advance to position them in the marketplace.
When is a Notified Body Required?
A Notified Body is typically required simultaneously as the product falls below a directive that mandates zero—33 birthday celebration evaluation, collectively with clinical gadgets, first-rate devices, or personal shielding gadgets. The Notified Body will conduct critical assessments and audits to ensure the product meets the necessities. For agencies in Abu Dhabi, deciding on a notified body that is diagnosed via the EU is critical to ensure the validity of the CE mark.
Working with Notified Bodies
Working with a notified body consists of several steps: submitting the product for testing, performing gift system audits, and presenting all critical documentation. The way can be time-consuming and calls for cautious coordination. However, the records of a Notified Body can help navigate the complexities of CE Certification in Abu Dhabi and ensure compliance.
Benefits of CE Certification for Abu Dhabi Businesses
Access to European Markets: The maximum benefit of acquiring CE certification in Abu Dhabi is gaining access to the European market. The EEA is one of the most critical and worthwhile international markets, and the CE mark certification in Abu Dhabi is a passport to this marketplace. For corporations in Abu Dhabi, this suggests the opportunity to make more significant profits, grow sales, and assemble a global presence.
Enhanced Product Quality and Safety: CE Mark Certification in Abu Dhabi ensures that products meet immoderate requirements of exceptionality and safety. This reduces the hazard of product recollects or criminal troubles and enhances the recognition of the emblem. Customers and business company companions in Europe and other regions recognize the CE mark Certification in Abu Dhabi as a photograph of reliability and compliance.
Competitive Advantage: In an aggressive market, having CE Mark certification in Abu Dhabi can set an agency apart. It demonstrates willpower to be remarkable and adhere to worldwide requirements, which may be a key selling detail. For companies in Abu Dhabi, this progressed client trust, stronger partnerships, and extra market percentage.
Conclusion
CE Mark Certification in Abu Dhabi is a critical step for groups in Abu Dhabi trying to expand into European markets and beyond. While the way may be complicated and challenging, the blessings outweigh the charges. By ensuring that products meet European necessities, agencies can enhance their recognition, access new markets properly, and attain prolonged fulfillment. Whether you’re a small or large organization, acquiring CE Mark certification in Abu Dhabi is an investment in the future that could release substantial growth opportunities.
Why Factocert for CE Mark Certification in Abu Dhabi?
We provide the best ISO consultants Who are knowledgeable and provide the best solution. And to know how to get CE Mark certification in Abu Dhabi. Kindly reach us at [email protected]. work according to ISO standards and help organizations implement CE Mark certification in Abu Dhabi with proper documentation.
For more information, visit CE Mark Certification in Abu Dhabi
Related Links
ISO Certification in Abu Dhabi
ISO 9001 Certification in Abu Dhabi
ISO 14001 Certification in Abu Dhabi
ISO 27001 Certification in Abu Dhabi
ISO 45001 Certification in Abu Dhabi
ISO 22000 Certification in Abu Dhabi
ISO 13485 Certification in Abu Dhabi
HALAL Certification in Abu Dhabi
CE MARK certification in Abu Dhabi
Related Articles:
Why is CE Mark Certification in Abu Dhabi Important
0 notes
Text
Schirmer Tear Test Strips: A Leap in Innovation and Certification by Ophtechnics Unlimited
Pioneering Quality and Compliance in Ophthalmology
At Ophtechnics Unlimited, we are proud to announce that we are the first company in India to achieve CE Certification under the EU MDR 2017/745 for our Schirmer Tear Test Strips. This milestone is not just a testament to our commitment to quality but also to our dedication to advancing ophthalmic diagnostics through rigorous compliance and innovation.

Advancing Standards with Schirmer Tear Test Strips
For ophthalmologists, precision and reliability in diagnostic tools are non-negotiable. Our Schirmer Tear Test Strips have been meticulously designed to provide consistent and accurate measurements of tear production, aiding in the diagnosis and management of dry eye syndrome and other ocular surface disorders. What sets our strips apart is not only their superior quality but also the rigorous testing and validation processes they have undergone to meet the stringent requirements of the EU MDR 2017/745.
The Journey to CE Certification
Achieving CE Certification under the new EU MDR 2017/745 regulations is no small feat. This regulation, which came into effect to ensure higher standards of safety and performance for medical devices, demands comprehensive documentation, rigorous testing, and thorough risk assessments. Our Schirmer Tear Test Strips have passed all these stringent checks, confirming their safety, efficacy, and reliability for clinical use.
Commitment to Excellence in Ophthalmic Strips
Our journey to CE Certification underscores our unwavering commitment to excellence. By adhering to the EU MDR 2017/745 standards, we ensure that our Ophthalmic Strips not only meet but exceed global standards. This dedication to quality is reflected in every aspect of our product development, from raw material selection to manufacturing processes, ensuring that ophthalmologists receive tools they can trust.
Global Impact and Reach
With our Schirmer Tear Test Strips now CE Marked, we are poised to make a significant impact on the global stage. Our products are already trusted by healthcare professionals in over 42 countries, and this certification opens new avenues for us to support ophthalmologists worldwide. By providing reliable and high-quality diagnostic tools, we aim to enhance patient care and improve outcomes in ophthalmology.
Conclusion
Ophtechnics Unlimited's achievement in obtaining CE Certification for our Schirmer Tear Test Strips under the EU MDR 2017/745 is a milestone in our journey towards innovation and quality. We remain dedicated to supporting ophthalmologists with the best tools available, ensuring precise diagnostics and effective patient care. As we continue to push the boundaries of excellence in ophthalmic strips, we invite you to join us in this exciting journey of advancing eye care globally. For more info visit us our website or contact us at [email protected] or +91-90-6262-3636
#ophthalmic surgical devices#ophthalmology#ophthalmic#eye surgical devices#ophthalmic medical instruments
0 notes
Text
Ensuring Compliance: Navigating CE Marking for Medical Devices
In the complex landscape of medical device regulation, obtaining the CE marking is a critical step for manufacturers looking to market their products in the European Economic Area (EEA). The CE marking indicates that a medical device complies with essential requirements outlined in the European Union's Medical Device Regulation (MDR). Navigating the CE marking process requires a thorough understanding of regulatory requirements, adherence to quality standards, and strategic planning. This article delves into the intricacies of CE marking for medical devices, highlighting key considerations and best practices for ensuring compliance.
Understanding CE Marking
The CE Marking for Medical Device is a symbol affixed to medical devices to demonstrate conformity with applicable European Union (EU) directives and regulations. It signifies that the device meets essential safety and performance requirements, enabling it to be legally marketed and distributed within the EEA. For medical devices, compliance with the Medical Device Regulation (MDR) is paramount.
Regulatory Framework
The MDR, implemented in May 2021, introduced stricter regulations for medical devices to enhance patient safety and ensure the effectiveness of these products. It outlines requirements for classification, clinical evaluation, post-market surveillance, and conformity assessment. Manufacturers must demonstrate compliance with relevant standards and undergo a rigorous assessment process to obtain the CE marking.
Classification and Conformity Assessment
CE Marking for Medical Device are classified into different risk categories based on their intended use and potential harm to patients. The classification determines the conformity assessment route, ranging from self-certification for low-risk devices to involvement of notified bodies for high-risk devices. Manufacturers must select the appropriate conformity assessment procedure and compile technical documentation to support conformity with applicable requirements.
Technical Documentation
The technical documentation serves as evidence of conformity with the MDR and includes detailed information about the design, manufacture, and performance of the medical device. It encompasses aspects such as product specifications, risk management, clinical evaluation, and labeling. Thorough and well-documented technical documentation is essential for a successful CE marking application.
Quality Management Systems
Establishing and maintaining a robust quality management system (QMS) is fundamental to achieving and maintaining CE marking for medical devices. Compliance with international standards such as ISO 13485 is commonly required to demonstrate the manufacturer's commitment to quality and regulatory compliance. Implementing effective QMS practices ensures consistency in product quality and regulatory compliance throughout the device lifecycle.
Clinical Evaluation
Clinical evaluation is a systematic assessment of clinical data to verify the safety and performance of a medical device. It involves gathering and analyzing clinical evidence to support the device's intended purpose and indications for use. Manufacturers must conduct clinical evaluations in accordance with the requirements specified in the MDR, considering factors such as equivalence, clinical investigations, and post-market surveillance data.
Post-Market Surveillance
Post-market surveillance (PMS) is an ongoing process aimed at monitoring the performance and safety of medical devices once they are placed on the market. It involves collecting and analyzing data from various sources, including adverse events, complaints, and feedback from healthcare professionals and patients. Effective PMS enables timely detection of potential issues and facilitates continuous improvement of product safety and performance.
Notified Bodies
Notified bodies play a crucial role in the CE marking process by assessing the conformity of medical devices with regulatory requirements. These independent organizations are designated by EU member states to perform conformity assessments for certain device categories. Manufacturers must engage notified bodies for conformity assessment of high-risk devices, involving rigorous scrutiny of technical documentation and quality systems.
Labeling and Packaging
Accurate and comprehensive labeling and packaging are essential for ensuring the safe and effective use of medical devices. Manufacturers must provide clear instructions for use, warnings, and precautions to mitigate risks associated with device use. Labeling should comply with regulatory requirements and standards, including language requirements for different EU member states.
Global Harmonization
While the CE marking is specific to the European market, achieving compliance with EU regulations can facilitate market access in other regions. Many countries outside the EU recognize CE marking as evidence of compliance with international standards and regulatory requirements. Manufacturers can leverage their CE marking certification to streamline regulatory approval processes in global markets, enhancing market competitiveness and expansion opportunities.
Conclusion
Navigating the CE marking process for medical devices requires a comprehensive understanding of regulatory requirements, diligent preparation, and commitment to quality and safety. Manufacturers must develop and implement robust quality management systems, compile thorough technical documentation, and undergo rigorous conformity assessment procedures. By ensuring compliance with the MDR and obtaining the CE marking, manufacturers can access the lucrative European market and demonstrate their commitment to delivering safe and effective medical devices to patients worldwide.
0 notes
Text
How can a Canadian company verify if a product meets CE Mark standards?
/ Uncategorized / By Factocert Mysore

What’s the Big Deal with CE Mark Certification in Canada ?
CE Mark Certification in Canada are very important when selling a product in Europe. If you’re a Canadian company, making sure that your product ticks all the boxes for these standards is a must. Let me walk you through how you can do that.
What are CE Mark Certification in Canada Standards?
A CE Mark Certification in Canada shows that a product passes Europe’s safety, health, and environmental checks. Things like electronics, machinery, medical devices, and toys need these marks. So, if you want to sell any of these in Europe, you’ve got to know your CE Mark Certification in Canada standards.
Which Rules Apply to Your Product?
First off, find out which European rules apply to your product type. Different products follow different rules. For example:
Medical devices have to follow the Medical Device Regulation (MDR).
Electrical equipment has to follow the Low Voltage Directive (LVD).
Machinery has to follow the Machinery Directive.
Assessing the Risk
The next part is all about risk assessment. Which means:
Identify possible dangers that come with your product.
Come up with ways to lessen these dangers.
Make sure your product has safety precautions built into it.
Following Harmonized Standards
Harmonized Standards are just CE Mark Certification Audits in Canada standards that help meet their essential directives. These Standards make it easier to pass the assessment process. You can get these Standards from the European Committee for Standardization (CEN) or the International Electrotechnical Commission (IEC).
Building a Technical File Next up:
Creating a technical file. This file proves your product meets the requirements. This file needs:
A thorough product description.
Drawings of design and manufacture.
Documentation of risk assessment.
Certificates and test reports.
Manuals for users and installation.
This file needs to be kept current and shown to European authorities when asked.
Conduct a Conformity Assessment
Conformity assessments can be done by the manufacturer. Sometimes, you might need a Notified Body to help. Notified Bodies are organizations picked by EU countries to make sure products meet the proper standards before they can be sold. They provide:
Independent tests and CE Mark Certification in Canada.
Checking that the technical file is complete and right.
Issuing a CE Mark Certification in Canada if the product passes the standards.
Declaring the Conformity
Once you’ve done the assessment, you have to create and sign an EU Declaration of Conformity (DoC). This document says the product passes all applicable EU checks and needs:
The maker’s name and address.
The product description.
Applied directives and standards.
Signature of a company’s representative.
Sticking the CE Mark Certification in Canada
After the DoC, the CE Mark Certification in Canada can be put on the product. The CE Mark Certification bodies in Canada needs to be:
Clear, visible and permanent.
Attached on the product, packaging, or documentation.
Along with the identification number of the Notified Body, if involved.
Keeping Current and Market
Monitoring you’re not done once the CE Mark is on. Canadian companies have to ensure they always comply by:
Updating the technical file if the product changes.
Doing regular checks and audits.
Fixing issues promptly if flagged by EU market surveillance authorities.
Use Available Resources and Knowledge
Canadian companies can pull from different sources to help with process:
Consult with EU regulatory experts.
Take part in CE Mark Certification in Canada training programs.
Collaborate with Notified Bodies for professional advice.
Conclusion
Checking if a product meets the CE Mark Certification in Canada standards is a thorough process with several important steps. Canadian companies have to understand EU rules, do hard-hitting risk assessments, stick to Harmonized Standards, and keep detailed technical documents. By doing this, companies can make sure their products pass the proper checks, making it easier to break into the European market. Regular updates and continuous compliance are the key to keeping certification and continued market access.
Why Factocert for CE Mark Certification in Canada
We provide the Best CE Mark Consultants in Canada who are knowledgeable and provide the best solution. And to know how to get ISO certification. Kindly reach us at [email protected]. work according to ISO standards and help organizations implement CE Mark Certification in Canada with proper documentation.
For more information, visit CE Mark Certification in Canada.
RELATED LINKS
ISO Certification in Canada
ISO 9001 Certification in Canada
ISO 14001 Certification in Canada
ISO 45001 Certification in Canada
ISO 27001 Certification in Canada
ISO 22000 Certification in Canada
ISO 13485 Certification in Canada
CE Mark Certification in Canada
0 notes
Text
Navigating the Transition: Understanding the UKCA Marking Certification Process for Medical Devices

The United Kingdom’s choice to leave the European Union has achieved tremendous changes, not just in that frame of mind of exchange and guidelines, but in addition to the accreditation processes governing the conformity of products. One pivotal part of this change is the entry of the UKCA (UK Conformity Assessed) marking, which replaces the CE marking for merchandise put on the UK market.
In this article, we will dig into the UKCA Marking in UAE certification process, giving bits of knowledge to assist organizations with exploring this basic change.
Scope and Applicability
Understanding whether an item requires UKCA marking is the most important phase in the certificate process. Not all products are dependent upon the new guidelines, and organizations should cautiously evaluate the particular necessities appropriate to their products. Certain items might in any case utilize CE marking, assuming that they are being sold in the EU and meet European guidelines.
What is UKCA Marking?
The United Kingdom Conformity Assessment Mark or UKCA Mark for short is what could be compared to the EU CE marking. UKCA Mark is a substantial pointer that a medical device adjusts to significant UK Guidelines. The United Kingdom Conformity Assessment marking is required for medical devices sold available in Great Britain (Britain, Wales, and Scotland).
United Kingdom’s Medicines and Health Care Products Regulatory Agency (MHRA) On 18 September 2019, distributed new rules to direct medical devices after Brexit. The UKCA won't be perceived in the EU, EEA, or Northern Ireland items actually require a CE marking available to be purchased in these business sectors.
The maker or their approved delegate will be answerable for fastening the UKCA Certification to the product, which is a similar rule concerning CE marking yet for the UK market.
What is UKCA Marking for Medical Devices?
The UKCA marking is a logo that implies a medical device's compliance with the UK MDR 2002 requirements. This mark certifies that the device is protected, fit for its planned reason, and conforms to the regulations connected with safety. It is a compulsory necessity for medical devices planned for the Great Britain market, which incorporates Britain, Wales, and Scotland.
Who Needs UKCA Marking for Medical Devices?
In the event that you are associated with the assembling, delivery, or import of medical devices in the UK, you might be expected to acquire UKCA marking. This incorporates:
Medical device makers situated in the UK
Merchants of medical devices in the UK
Organizations importing medical devices into the UK
Technical Documentation and Standards
The core of the certificate process lies in the preparation & accommodation of technical documentation. Makers should order an exhaustive record framing the item's plan, details, and conformity with significant UKCA Marking standards. Experience with these norms is significant, and organizations might have to adjust their cycles to meet the particular measures set by UK specialists.
Documentation Required for UKCA Certification:
Records
Formats
Forms
Checklist
Standard Operating Procedure (SOP)
Plant Master File
Mission & Vision
Objectives
Policy
System Procedure
System Manual
Product Testing
Technical File, Product Master File (TCF)
The extent of Documented Information differs as per:
Notify body Certificate
Self Certification or Compliance Certification
Directive in which product classified
Testing requirements of products
Product and its uses
Key Requirements for Placing Medical Devices in the Great Britain Market:
New Product Marking (UKCA Mark): Guarantee that your medical device delivers the UKCA mark.
Registration with MHRA:: Every single medical device and IVD should be enrolled with MHRA before they can be made available.
UK Responsible Person: In the event that you are a producer situated external the UK, you should select a single UK-reliable individual.
Classification: Medical devices in the UK are classified as Class I, Class IIa, Class IIb, and Class III, like the European Union’s plan. Class I devices are generally safe, while Class III devices are high-risk.
Registration Process for Medical Devices (UKCA Marking Process)
The Medicines and Healthcare Products Regulatory Agency is the UK's regulatory power answerable for guaranteeing that meds and medical devices are protected and compelling.
Classification: Decide the order of your medical device as indicated by the MDR.
Conformity Assessment: Lead a conformity evaluation to show that your device meets security and execution requirements.
Specialized Documentation: Get ready tech documentation that incorporates device depictions, planned use, and medical information.
Application Submission: Present your application to the MHRA through the Medical Devices Information System (MDIS).
Assessment: MHRA will assess your application and specialized documentation to guarantee compliance with safety and execution prerequisites.
Registration Certificate: In the event that your application is effective, MHRA will give an enrollment declaration, permitting you to sell your medical device in the UK.
Additional Points:
A progress period exists for certain item products, permitting the utilization of existing CE marking until Dec. 12, 2027.
The particular process can shift contingent on your item type and intricacy.
Conclusion: A Roadmap for Compliance
Exploring the UKCA Marking in UAE certification process requires a key and informed approach. Organizations should put time and assets into understanding the subtleties of the new regulatory scene, guaranteeing that their items fulfill the expected guidelines for the UK market.
As the business climate keeps on developing, remaining adaptable and receptive to changes will be vital to keeping an upper hand and guaranteeing continuous admittance to the UK market. By embracing the UKCA marking process for medical devices as a chance for development and compliance, organizations can unhesitatingly explore the change and secure their situations in the post-Brexit time.
0 notes
Text
Switzerland Authorised Representatives | Swiss Medical Device Regulations | OMC Medical

Switzerland Medical Device Regulations
Medical devices are regulated: Swiss Agency for Therapeutic Product (Swissmedic)
Current Switzerland Market Situation
As the Institutional Agreement is missing (InstA) is missing, the EU has not updated the Mutual Recognition Agreement(MRA)
No barrier free access to the EU internal market.
Swiss has been downgraded to the third country.
Swiss manufactures has stricter requirements when exporting their medical devices to the EU.
Manufacturer Responsibility
The center of the responsibility defined by law.
Prove and document conformity of his product, including correct classification.
Introduction and maintenance of a QMS system is his responsibility.
AR is dependent on the manufacturer to fulfil his obligations.
Manufacturer must be contractually obliged to guarantee the authorized representative full compliance of his legal duties.
Legal duties include- CE mark, UDI, IFU, Translation into Swiss languages.
Manufacturer must supply the AR with extensive documentation.
Manufacturer is obliged to retain the technical documentation, declaration, certificate of conformity, including amendments and supplements for 10-15 yrs.
AR to keep commercial records for 10 yrs.
Manufacturers outside Swiss
After 26 May 2021 , foreign manufacturers must appoint a Swiss authorised representative in order to sell their devices in the market.
Agreement between the manufacturer and the AR is needed.
Who is a Swiss AR
– Proxy to the foreign manufacturer – Responsible for product safety – Liable for product defects – Contact person for Swiss authorities – Can be legal entity or as a natural person – Must have access to PRRC
Swiss Authorised Representative
A Swiss authorised representative (Swiss AR) is a legal person appointed by a manufacturer established outside of Switzerland to place the products on the Swiss market.
A written mandate/letter designating an authorised representative for a medical device manufacturer is mandatory.
Manufacturers can appoint only a single Authorised representative in Switzerland.
According to the Swiss ordinance on medical devices, the authorized representative is responsible for any product defects. AR is jointly and severely liable for product defects with manufacturer.
Update for Manufacturers in EU & Swiss
Registration and notification obligations do not run via EUDAMED, but via Swissmedic.
Economic operators who have already placed products on the market before 26 May 2021 in accordance with the MDR and IVDR must complete their registration by 26 November 2021.
Access to the technical documentation may be provided either by keeping a copy available at the authorised representative or by contractually guaranteeing that it will be handed over to Swissmedic upon request within 7 days.
The validated Summary of Safety and Clinical Performance (SSCP) is not uploaded by the Notified Body in EUDAMED, but published by the manufacturer, for example on its website.
EU Manufacturer Exporting to Swiss
Need to appoint Swiss AR, update the labelling for MDR products & follow third country requirements.
Timeline to appoint Swiss AR:
Swiss Manufacturer Exporting to EU
For MDR regulated devices & class I devices: Follow third country requirements from 26th May 2021.
For MDD certified devices: No third country requirements, they will be regulated under MRA, free access to the EU market as before until May 2024.
How we assist you with this process?
Act as your Swiss Authorised Representative
MedDO compliance
EUDAMED registration
Product information translations in German, French, Italian language
EU MDR, IVDR gap analysis
Why Choose Us?
Working towards client satisfaction
Cost effective solutions
Project completion before deadline
Quality Regulatory affairs solutions
Originally Published at: https://omcmedical.com/switzerland-ar/
0 notes
Text
CE Mark Certification Consultants in Saudi Arabia
What Is the European CE Mark Standard for Medical Devices?
CE mark for medical devices in Saudi Arabia means a symbol with the acronym ‘CE’ on the product, which is proof that the manufacturer of medical devices is taking care of all regulatory requirements. As per the EU MDR, CE marking is required for medical devices /products to be legally placed in the EU market.
0 notes
Text
EU MDR compliance for Legacy Devices
EU MDR compliance for Legacy Devices
Legacy devices are the medical devices, active implantable medical devices and in vitro diagnostic devices that are placed in the market with valid certificates issued from Medical Devices Directive (MDD) or Active Implantable Medical Device (AIMD). The valid CE (Conformité Européenne) marking certificates are issued according to Directive 93/42/EEC, Directive 90/85/EEC or Directive 98/97/EC on medical devices. The legacy devices continue to be in market when it meets EU MDR compliance for legacy devices as per set timelines.REGISTRATION OF LEGACY DEVICESA document “MDCG 2019-5 “Registration of legacy devices in EUDAMED April 2019” explains the guidance on registration of legacy devices in EUDAMED. EUDAMED is the European Database for Medical Devices. According to new guidelines the legacy devices are registered in EUDAMED. The registration of legacy devices is mandatory when there is a serious incident related to the product or there is any field corrective action necessary towards it.A NEW GUIDANCE ON EU MDR COMPLIANCE FOR LEGACY MEDICAL DEVICESThe European Commission’s Medical Device Coordination Group (MDCG) ‘s has put forth a new guidance that describes the implementation and application of MDR requirements for legacy devices and the devices placed in the market before 26 may 2021. The MDGC had set up ad hoc task- force for applying transitional provisions to the legacy devices and devices placed in the market before 26 may 2021 mentioned in Article 120(3) of Regulation (EU) 2017/745 (MDR)Read More - EU MDR compliance for Legacy Devices Read More Article - 1. Orthopedic Implants Manufacturing 2. disposable syringe manufacturers 3. Dental Implants Manufacturing 4.Blood Collection Tubes Manufacturing Contact details – Phone no - 93702 83428Mail id – [email protected]
#mdr compliance#eu mdr compliance for legacy devices#mdr requirements for legacy devices#eu mdr ce marking certification for medical devices#eu mdr compliance#registration of legacy devices#new identifiers for the legacy devices#a new guidance on eu mdr compliance for legacy medical devices#mdr requirements to legacy devices
0 notes
Text
Design to Sale regulatory assistance - We provide high quality work with low fees comapred to competitive rates in the market. UKRP, Regulatory Intelligence, Regulatory country submissions, Labelling and Art work, Global Language translation, SaMD (Software as a Medical Device), Regulatory strategy, Design to Sale regulatory assistance, Regulatory Training, Database Maintenance, Regulatory staffing-Interim and Permanent. https://omcmedical.com/services/
#Labelling and Art work#CE marking#Cosmetic Registration#EUDAMED#UKCA#UK Responsible Person#UKRP#Medical Device Registration#Local EU listing#Inmetro certification#EU MDR#Clinical Evaluation#Regulatory Strategy#Regulatory Training
0 notes
Link
Freyr provides regulatory support for medical device manufacturers in preparation of Clinical Evaluation Report (CER), which includes report writing, GAP analysis, Post-market Surveillance(PMS) data support for existing devices and updating Clinical Evaluation Report (CER) as per EU MDR guidelines/regulations.
0 notes
Text
Steps for Getting CE Marking
All products meeting the definition of medical devices as defined by the EU regulations require CE marking before they can be sold in the EU. CE marking indicates that a product has been assessed by the manufacturer and meets EU safety, health, and environmental protection requirements. It is required for products manufactured anywhere in the world that are then marketed in the EU.
The product must comply with all the relevant MDD to MDR requirements of before affixing the CE marking to it. Although the compliance requirements are similar in many ways, the European route is thought to be less governmental, resulting in shorter times to market, greater acceptance rates of new devices, and lesser costs associated with obtaining conformity certification.
While the benefits of obtaining a CE marking are significant, regulations are always subject to change. New modifications to the European medical device requirements are making the process more similar to that of the FDA when it comes to establishing conformity.
By May 2021, a new Medical Device Regulation (MDR 2017/745) will go into effect throughout the European Union. Despite the changes, there still remains a clear path to establishing conformity and obtaining a CE marking that will allow your company to access European markets.
Steps
1. IDENTIFY THE APPLICABLE REQUIREMENTS OF THE REGULATIONS
1. CLASSIFY THE DEVICE AS PER THE RULES DEFINED FOR CLASSIFICATION OF MEDICAL DEVICES
2. IDENTIFY AN APPROPRIATE ROUTE TO CONFORMITY
3. ASSESSMENT OF THE PRODUCT’S CONFORMITY TO EU REQUIREMENTS
4. COMPILE THE TECHNICAL DOCUMENTATION, QMS & CLINICAL EVALUATION REPORT.
5. ASSESSMENT BY NOTIFIED BODY
6. MAKE A DECLARATION OF CONFORMITY AND AFFIX THE CE MARK
IZiel Healthcare has collaborated with Belgium based Obelis (European Authorized Representatives) to provide a “One-Stop Solution” to fully support Class I, IIa, IIb & III medical device manufacturers across USA, Europe & Asia. Typically, all MDD-MDR transition projects initiate with Gap Assessment. Gap Assessment is a crucial activity during MDR transition.
IZiel-Obelis collaboration would ensure to obtain conformity with the MDR (2017/745) requirements and maintain the CE Marking by developing Technical File & QMS Documentation, conducting Software Validation, writing CERs and providing EC Rep, EUDAMED, PRRC Services. Our experts are well equipped to conduct this activity with an analytical mindset, resolve any engineering requirements and develop robust regulatory strategy for medical device manufacturers.
#regulation#ce marking certification#usfda#processvalidation#technical design document#software validation
1 note
·
View note
Link
Technical File for medical device is a must for all type of devices regardless of the risk classification. Manufacturers are advised to start working on revising the CE Marking technical regardless of Notified Body.
New European medical device regulation (MDR) and In Vitro Diagnostic Regulation (IVDR) the CE Certificate will be issued by Notified Body after a thorough review of Technical File Documentation.
The File must be prepared and submitted to notified body by the manufacturer.
The new (EU MDR 2017/745) Medical Devices Regulation and (IVDR 2017/746) In Vitro Diagnostic Regulation has a lot of additional requirements compared to medical device directive which is explained below.
1 note
·
View note
Text
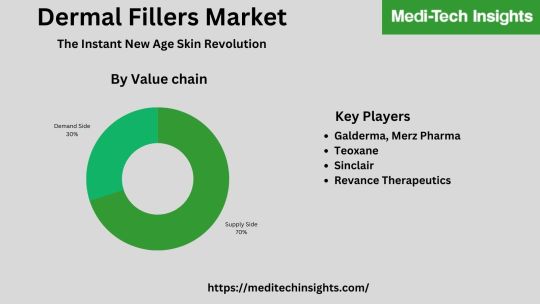
Global Dermal Fillers Market was valued at US$ 2.8 billion in 2021 and is likely to grow at a CAGR of 10% by 2026
Dermal fillers are gel-like substances that are injected under the skin to restore lost volume, smooth lines and soften creases, and enhance facial contours.
The Global Dermal Fillers Market was valued at US$ 2.8 billion in 2021 and is set to witness a healthy growth rate of 10% in the next 5 years. ‘Covid-19-Zoom-Boom Effect’, an growing influence of social media in promoting non-surgical aesthetic procedures, rising focus on physical appearance, increasing cultural acceptance & fading social stigma on cosmetic procedures, increasing demand from younger population and men, rise in the number of clinics offering non-surgical aesthetic services, expanding geriatric population & rising awareness about anti-aging cosmetic procedures are some of the key factors driving the global dermal fillers market.
Covid-19, Financial Bottlenecks, 'Zoom Boom‘ and Rebound of the Dermal Fillers Market
Covid-19 had a negative effect on the dermal fillers market globally, forcing a number of aesthetic service providers to alter the way they delivered their services. Facial aesthetics clinics were not allowed to operate, and people were not allowed to visit them. For a while, business was entirely shut down, which had a negative financial impact on the aesthetics industry. These disruptions encouraged several suppliers to advance their businesses to online platforms and ordering systems.
However, the aesthetics industry is currently on the path to recovery despite the Covid-19 pandemic. Since early 2020, aesthetics clinics worldwide have witnessed an overwhelming spike in bookings for cosmetic enhancements due to Covid-19 & the ‘Zoom Effect. It's been dubbed the "Zoom Boom" in the sector because people are using video calls more for work and socializing and hence have become more conscious of their appearance after seeing their faces (facial expressions & wrinkles) on video calls every day on Zoom. Since 2020, demand for consultations with plastic surgeons has increased exponentially, according to reports from across the globe.
Regulation of Dermal Fillers Likely to Become Stricter in the European Union (EU) With the Introduction of New Medical Device Regulation (MDR)
Within the EU, medical devices used to be regulated through the Medical Devices Directive (MDD), which required all medical devices to have a CE certification. This did not include dermal fillers.
However, from May 2021, the MDD has been replaced by MDR which classifies dermal fillers as Class III medical devices. As a result of the new MDR, manufacturers must comply to its requirements. If manufacturers fail to comply with the MDR requirements, they will be unable to market their products within the EU.
“The EU move to MDR is likely to create entry barriers. Smaller manufacturers who cannot support their products with clinical studies will be driven out.”- Head of Aesthetics Division, Leading Aesthetic Product Manufacturer, UK
Explore Premium Report on Dermal Fillers Market @ https://meditechinsights.com/dermal-fillers-market/
Organic and Inorganic Growth Strategies Adopted by Players to Establish Their Foothold
The dermal fillers market is marked by the presence of both established and new players. Players operating in the market adopt both organic and inorganic growth strategies such as new product launches, and partnerships to garner market share.
For instance,
In December 2021, Revance’s partner, Teoxane SA, received U.S. FDA approval for RHA® Redensity’s first indication, which is for the treatment of moderate to severe dynamic perioral rhytids (lip lines) in adults aged 22 or older. RHA® Redensity bolsters the versatility of the RHA® Collection of dermal fillers
Due to the rising importance of digitalization, automation tools, and data-driven solutions that make use of predictive analytics, AI, and ML, the dermal fillers market is expected to see double-digit growth in the upcoming years.
These tools are being utilized in increasing numbers for:
Streamlining resource allocation in medical facilities
Doctors and care managers can outline populations, target their most complex patients, and identify rising-risk patients
Managing patient load and assisting in capacity management - Allocating emergency rooms based on the severity of the illness, and reducing patient service turnaround time
Streamlining the insurance claim process that costs the hospitals significant labor hours. The technology also assists in checking eligibility and subsequent data migration, quickly and effectively
Competitive Landscape Analysis of Dermal Fillers Market
The global dermal fillers market is marked by the presence of players such as Allergan (Part of AbbVie), Galderma, Merz Pharma, Teoxane, Sinclair, Revance Therapeutics, Prollenium Medical Technologies, Bioxis Pharmaceuticals, among others.
For More Detailed Insights, Contact Us @ https://meditechinsights.com/contact-us/
About Medi-Tech Insights
Medi-Tech Insights is a healthcare-focused business research & insights firm. Our clients include Fortune 500 companies, blue-chip investors & hyper-growth start-ups. We have completed 100+ projects in Digital Health, Healthcare IT, Medical Technology, Medical Devices & Pharma Services.
Contact:
Ruta Halde
Associate, Medi-Tech Insights
+32 498 86 80 79
0 notes