#mdr compliance
Explore tagged Tumblr posts
Visit Tumblr Blog
Explore Tumblr blogs with no restrictions, modern design and the best experience.
Last Seen Tumblr Blogs





Fun Fact
Tumblr has 16.74 million mobile monthly users in the US.
Text
For manufacturers of in vitro diagnostic (IVD) devices seeking to market their products in the European Union, ensuring MDR compliance is a critical requirement. The EU Medical Device Regulation (MDR) has brought about significant changes in the regulatory landscape, and manufacturers must navigate these new rules to achieve and maintain compliance. Before divulging details, it is essential to understand IVD compliance.
0 notes
Text
1 note
·
View note
Text
Medical Device Labeling: The Important Role Of Medical Device Label In Patient Care

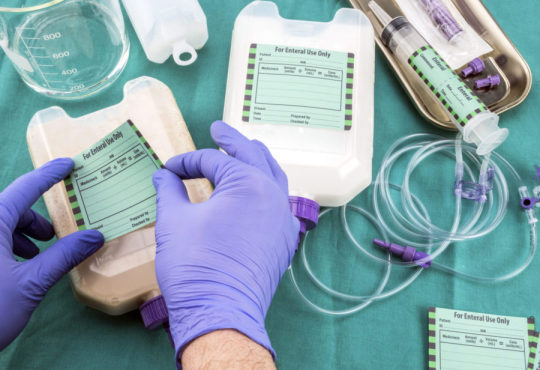
Medical devices sold in the United States must comply with labeling requirements set by the Food and Drug Administration (FDA). All labels must contain specific information about the device such as its intended use, any potential risks or side effects, and instructions for use. Device labels provide critical safety information that helps ensure devices are used properly. The FDA regulates labeling to protect patient health.
Labels Must Clearly Identify The Device
First and foremost, a Medical Device Labeling must clearly identify the specific device. This includes stating the product name and any applicable product codes or reference numbers. Having a clear device name and identification numbers helps providers and facilities properly select, use, and track the intended device. Any trade or brand names must also be included but must not overshadow the core product identity information.
Instructions For Use Must Be Clear And Complete
Detailed instructions for use are extremely important for medical devices to be operated safely and as intended. Labels must provide step-by-step directions on how the device is to be used, prepared, fitted, applied, implanted, or operated. Pictures, diagrams or other illustrative guides can help visualize proper technique when words alone may not fully explain the process. Instructions must account for all reasonably foreseeable uses. Omitting any necessary steps could result in misuse leading to patient harm.
Packaging Labels Provide Sterility And Shelf Life Assurances
For devices distributed in single-use sterile packaging, labels must affirm the method used to sterilize the contents as well as the expiration date or shelf life. This information guarantees the continued sterility and integrity of the device up until the noted expiration date. Devices like surgical tools and implants must remain free of contaminants when used. Packaging labels demonstrate the steps taken to achieve and maintain sterile conditions.
Potential Adverse Reactions And Hazards Must Be Disclosed
Device labels must include a complete list of known or reasonably foreseeable adverse health effects or hazards from use. This involves describing any potential allergic reactions, biological risks, toxicology concerns and interactions with other devices, drugs or substances. Precautions, contraindications and any use limitations due to patient risks or conditions should also be detailed. Making providers aware of safety issues enables them to properly assess risks and benefits for individual patients.
Symbols Standardize Hazard And Safety Communications
Pictograms and symbols play an important role in medical device labeling by allowing concepts to be recognized universally without language barriers. Common symbols indicate requirements such as “Do Not Reuse”, “Sterilized Using Irradiation”, “Keep Away From Heat or Flames”, and many others defined by International Standards Organization (ISO) regulations. Having standardized symbols helps labeling information be consistently interpreted.
Product Labels Remain With The Device
Device manufacturers must ensure all labeling remains affixed or adjacent to the medical device itself throughout distribution and use. Shipping labels serve to identify devices during transportation but do not replace the requirement for complete labeling on the finished able product. Device labels need to be available anytime and anywhere the device is utilized to provide critical use and safety guidance to providers.
As described, medical device labelingserves the fundamental purpose of guiding appropriate and safe use while communicating potential risks. Complying with comprehensive FDA labeling policies supports quality patient care.
Get more insights on this topic: https://www.pressreleasebulletin.com/medical-device-labeling-device-labeling-regulations-ensuring-patient-safety-and-compliance/
About Author:
Ravina Pandya, Content Writer, has a strong foothold in the market research industry. She specializes in writing well-researched articles from different industries, including food and beverages, information and technology, healthcare, chemical and materials, etc. (https://www.linkedin.com/in/ravina-pandya-1a3984191)
*Note: 1. Source: Coherent Market Insights, Public sources, Desk research 2. We have leveraged AI tools to mine information and compile it
#Medical Devices#Regulatory Compliance#Product Labeling#FDA Regulations#UDI (Unique Device Identification)#MDR (Medical Device Regulation)#Labeling Standards#Risk Management#Health Canada Regulations#Clinical Labeling
0 notes
Text
lying in bed awake thinking about the claymation video that milchick shows the mdr team about the microdat uprising. like it’s the fact that the characters and the (fictional) violence they enact is being represented in a visual medium typically used for children’s cartoons, which speaks to lumon’s infantilizing tone towards innies as a means of encouraging their compliance while denying their autonomy… it’s the claymation puppets as mirrors of the mdr team, who are also being manipulated into performing the leading role in lumon’s narrative…it’s the depiction of the lumon building as an anthropomorphic cartoon character, which means that the innies are once again being denied any access to information about the outside world and must depend on lumon’s representation of reality
#severance season 2#severance spoilers#i loveeeee thinking about how medium can inform narrative especially when it’s an embedded story that contests the main narrative#severance#this show has me foaming at the mouth yall
96 notes
·
View notes
Text
youtube

MDR 2017/745: Medical Device Labeling Requirements
Explore the labeling requirements for medical devices under MDR 2017/745. Learn what’s needed for compliance, from safety info to product details.
#labeling#Labeling_requirements#MDR2017#Medical_Device_Regulation#euregulations#regulatorycompliance#regulatory_solutions_india#Youtube
0 notes
Text
ISO 13485 Certification: A Gateway to Quality in the Medical Device Industry
Introduction In the highly regulated medical device sector, ensuring product safety and efficacy is paramount. ISO 13485 certification stands as a globally recognized benchmark for quality management systems (QMS) tailored to this industry. This article explores the significance of ISO 13485, its benefits, the certification process, and strategies to overcome implementation challenges.
What is ISO 13485? ISO 13485 is an international standard (latest version: ISO 13485:2016) that specifies requirements for a QMS designed explicitly for medical device manufacturers. Unlike ISO 9001, which applies broadly to quality management, ISO 13485 emphasizes regulatory compliance, risk management, and traceability—critical factors in medical device production. It is harmonized with regulations such as the EU Medical Device Regulation (MDR) and is often a prerequisite for market access in regions like Europe, Canada, and Australia.
Why is ISO 13485 Certification Important?
Regulatory Compliance: Many countries require iso 13485 certificering for market entry. For example, the EU links it to CE marking, while Health Canada mandates it for device licensing.
Enhanced Product Quality: Streamlined processes reduce errors, minimize recalls, and ensure consistent compliance with customer and regulatory requirements.
Global Market Access: Certification demonstrates commitment to quality, easing partnerships with distributors and healthcare providers worldwide.
Competitive Edge: Differentiates companies in tenders and builds trust with stakeholders.
The Certification Process: A Step-by-Step Guide
Gap Analysis: Assess current QMS against ISO 13485 requirements to identify gaps.
QMS Development: Establish policies, risk management protocols, and documentation (e.g., procedures, work instructions).
Implementation: Train employees and integrate QMS into daily operations.
Internal Audit: Conduct audits to verify compliance and address non-conformities.
Management Review: Ensure QMS effectiveness and readiness for external audit.
Certification Audit (Two Stages):
Stage 1: Documentation review by an accredited body.
Stage 2: On-site audit evaluating QMS implementation.
Surveillance Audits: Annual checks to maintain certification, with full recertification every three years.
Challenges and Solutions
Complex Documentation: Use digital QMS tools to streamline record-keeping and ensure traceability.
Regulatory Updates: Monitor agencies like the FDA or EU MDR through subscriptions or consultants.
Employee Resistance: Foster a quality culture with ongoing training and leadership buy-in.
Cost and Time: Small businesses can adopt phased implementations or seek government grants.
Conclusion ISO 13485 certification is more than a compliance checkbox—it’s a strategic investment in quality and reliability. By aligning with this standard, medical device companies not only meet regulatory demands but also enhance operational efficiency and global competitiveness. While challenges exist, proactive planning and resource allocation can smooth the path to certification, ultimately safeguarding patient health and corporate reputation.
Final Thoughts As the medical device industry evolves, ISO 13485 remains a cornerstone of excellence. Whether you’re a startup or an established player, achieving this certification underscores your commitment to delivering safe, effective products in an ever-changing regulatory landscape.
0 notes
Text
CE Marking: Guide for Manufacturers Seeking EU Market Access
CE marking, short for "Conformité Européenne" (French for "European Conformity"), is a mandatory symbol that indicates a product complies with the European Union's health, safety, and environmental protection standards. This marking is essential for products sold within the European Economic Area (EEA). By affixing the CE mark, manufacturers declare that their products meet all applicable EU directives and regulations, allowing for the free movement of goods across EEA countries. It's important to note that the CE mark is not a quality indicator or certification mark; rather, it signifies legal compliance with EU legislation.

The CE marking process involves several key steps to ensure that a product complies with European Union (EU) regulations and can be legally sold within the European Economic Area (EEA). Here's a step-by-step breakdown:
1. Identify Applicable Directives and Regulations:
Determine which EU directives or regulations apply to your product. Each directive outlines specific requirements related to safety, health, and environmental protection. For example, medical devices are governed by the Medical Devices Directive (MDR 2017/745).
2. Assess Specific Requirements:
Understand the essential requirements stated in the applicable directives. This may involve conducting a risk analysis and ensuring your product meets performance and safety standards.
3. Determine the Conformity Assessment Procedure:
Decide whether your product requires assessment by a Notified Body—a third-party organization authorized by the EU to evaluate product conformity. Not all products need this; some can be self-assessed by the manufacturer.
4. Conduct Product Testing:
Perform necessary tests to verify that your product meets EU requirements. Depending on the product and applicable standards, this testing can be done by an ISO 17025 accredited laboratory.
5. Compile Technical Documentation:
Gather comprehensive documentation detailing your product's design, manufacturing process, and compliance with relevant standards. This technical file should include:
Product description
Design and manufacturing drawings
Risk assessments
Test reports
User manuals
Declarations of conformity
6. Draft and Sign the EU Declaration of Conformity:
Prepare a document in which you, as the manufacturer or authorized representative, declare that the product complies with all relevant EU directives. This declaration must include:
Manufacturer's name and address
Product details (e.g., model, type)
List of applicable EU directives
Standards used to verify compliance
Date and place of issuance
Name and signature of the responsible person
7. Affix the CE Marking:
Once all the above steps are satisfactorily completed, affix the CE marking to your product. Ensure the marking is:
Visible, legible, and indelible
At least 5 mm in height (unless specified otherwise)
Proportional if resized If a Notified Body was involved, include their identification number alongside the CE mark.
By following these steps diligently, manufacturers can ensure their products meet EU standards and are eligible for sale within the EEA.
Why Choose NexorTest Technologies for CE Marking Certification?
We simplify the CE marking process with step-by-step guidance.
Our experts identify the right EU directives and standards for your product.
Save costs with our in-house accredited testing lab.
Tailored solutions ensure your product meets all CE requirements.
From compliance checks to affixing the CE mark, we manage it all.
Our streamlined process saves time and eliminates complexity.
Ensure EU regulatory compliance with precision and confidence.
Seamless market access and enhanced product credibility guaranteed.
Gain a competitive edge with our reliable support.
Contact NexorTest Technologies today to achieve hassle-free CE certification!
#ce marking#ce marking certifications#nexortest technologies#nexortest bengaluru#eu market ce certification
1 note
·
View note
Text
ISO 13485 Certification Consultants: Simplifying Compliance to Unlock Expertise Requirements
ISO 13485 is the international standard for Quality Management Systems in the manufacturing of medical devices, so this is a strategic initiative for organizations to guarantee product quality, comply with regulatory requirements, and boost trust in the market. However, understanding the complexities of ISO 13485 requirements can be a challenge.
This is where ISO 13485 Certification Consultants come in handy, bringing in deep expertise and insights to smoothen the certification process.
This page reviews how ISO consultants rely on their knowledge to assist organizations in meeting ISO 13485 requirements with services that include gap analysis, document preparation, training, and system deployment.
Thorough Understanding of ISO 13485 Standards
ISO 13485 in UAE is a strict set of requirements for a quality management system applicable to the medical device industry. It is focused on risk management, process control, and adherence to regulatory obligations.
Consultants become well-versed in these standards and how they should be applied, and they help organizations to:
Read the clauses of the standard correctly.
ISO 13485 process mapping and gap analysis
Verify adherence to relevant regulatory guidelines such as FDA and EU MDR.
Their knowledge eliminates confusion and affirms a clear roadmap for certification.
Conducting a Gap Analysis: Where Are You Non-Compliant?
This ensures that adjustments are well-informed and based on an understanding of the organization's current state relative to ISO 13485 requirements.
a. Mapping Processes to the Standard: The consultant undertakes a thorough analysis of all operational areas, including design controls, production processes, supplier handling, and post-market surveillance, and points out aspects that require improvement.
b. Prioritizing Actions: The gap analysis identifies high-priority items to fix, allowing work to be targeted and efficient to meet any regulations.
Training also requires data on documentation development and review
ISO 13485 Certification in UAE requires a lot of documentation and this can be daunting for organizations that do not understand how the standard is designed and how its requirements interrelate. Consultants offer customized support to develop and improve:
a. Quality Manuals: They write or update quality manuals to document organizational objectives, purpose, and compliance approaches.
b. How SOPs Should Be Maintained: Consultants make sure that SOPs are not only accurate and consistent but also compliant with ISO 13485.
c. Records and Reports: They oversee the preparation of critical documents such as risk assessments, nonconformance reports, and corrective action logs, making sure they adhere to audit requirements.
Risk Management Expertise
Risk management is one of the main components of the standard. ISO 13485 Consultants in Dubai assist organizations in assimilating risk-based thinking in their procedures by:
a. Developing Risk Assessment Frameworks: They develop frameworks for assessing, analyzing, and managing risk across product design, manufacturing, and post-market operation.
b. Perform Failure Mode, Effects, and Criticality Analysis (FMECA): By guiding teams through FMEA processes, consultants help identify risks before they occur and make products safer.
Learning and Development
ISO 13485 is a collaborative process for the whole organization. Consultants produce customized training courses to give staff members the skills and knowledge and skills they want.
a. Staff Training: Training on the ISO 13485 requirements for employees at all organizational levels consists of:
Principles of Quality Management
Documentation processes.
Risk management techniques.
b. Internal Auditor Training: The internal auditors learn to assess compliance through the guidance of consultants, preparing them for external audits.
Implement and Integrate System
Consultants in Abu Dhabi help with designing systems based on the needs of the organization, which include:
a. Process Optimization: They automate workflows to guarantee consistency, traceability, and adherence to regulatory standards and guidelines.
b. Software Solutions: Consultants recommend and implement QMS software tools to make document control, training management, and risk analysis easier.
c. Supplier Management Systems: They are the ones who are responsible for establishing strong systems to collect and track whether suppliers and their products are up to standard and fulfilling the regulations across the supply chain.
Pre-Audit Preparation
ISO 13485 standard audit preparation One of the significant functions of ISO 13485 Consultants is to prepare organizations for a certification audit. Their support includes:
a. Mock Audits: Mock audits are performed by consultants to mimic actual certification evaluations, in which weaknesses and opportunities for improvement are identified.
b. Audit Documentation: They guarantee that all necessary documentation is available at their fingertips, organized as needed, and in line with what the auditor may expect.
c. Staff Preparation: Trainers teach employees how to face auditor questions with confidence and accuracy.
Post-Certification Support
Having ISO 13485 is not the finale of a journey. Continual compliance support from consultants such as:
a. Continuous Improvement: They assist organizations, from time to time, to create processes to ensure QMS (quality management system) performance has been analyzed and improved accordingly.
b. Re-Certification Assistance: When ISO 13485 certificates must be renewed, consultants help organizations manage the re-certification process to ensure that it goes as smoothly as possible.
c. Changes to the Updates to the Standards: ISO 13485 consultants provide continuous information related to amendments with ISO 13485 to associations so they stay in line with the current necessities.
Benefits of Hiring ISO 13485 Certification Consultants
The consultants specializing in ISO certification provide substantial benefits through expertise, such as:
Less time to gain certification.
Reduced risk of non-compliance and audit failures.
Increased efficiency and quality control in the system.
Improved staff performance and motivation.
The Bottom Line!!
ISO 13485 standard reflects an organization's dedication to quality, safety, and regulatory compliance in the medical device sector. Nevertheless, certification is both complex and can be difficult to maintain when you lack the right know-how.
ISO 13485 Certification Consultants in UAE are indispensable allies in this process, offering targeted expertise, tailored advice, and practical roadmaps that simplify the certification journey. They provide comprehensive support throughout the entire process, from documentation and training to audit preparation and post-certification assistance, ensuring that organizations not only achieve ISO 13485 compliance but also develop strong systems for long-term success.
Therefore, working with an ISO consultant to ensure that a sound quality management system is in place will not only give you peace of mind but also save you time and money in the long run.
0 notes
Text
Identifying Potential Problems: CE Marking Certification Consultants Help Resolve Issues Early

CE marking is one of the most significant signs for producers ensuring that goods adhere to the European Union requirements of security, well-being, and natural standards. The CE mark is more than a piece of paper; it ensures quality and compliance with global standards.
Nonetheless, obtaining CE marking standards is not always easy, and many companies face the complexities of the process. This is where CE Marking Certification Consultants come into play.
They’re critical for enabling manufacturers to detect and correct potential problems early on and ensure that the product is compliant with all regulations before it hits the market.
The Guide to CE Marking Certification Process
CE Marking in UAE: A mandatory conformity mark for certain consumer goods in the European Economic Area (EEA) and a declaration that the product meets the safety and environmental protection standards of the EU. Earning CE marking certification is typically a multi-stage process, which includes product testing, risk assessment, technical documentation, and conformity assessment.
This process can be burdensome for manufacturers, as it demands extensive compliance with various regulations and a multitude of technical requirements.
It is where professionals like CE consultants come in. This is where experts come into play, they coach the companies through the certification process, making them realize the requirements, preventing any financial loss, and helping them with compliance.
How CE Marking Certification Consultants Add Value
A Guide to the NFC Implementation Process
Manufacturers are first shocked with detailed and frequently complex regulatory requirements when searching for CE marking in UAE depending on the product, industry, and even country in the EU these regulations vary. Some products are held to well-defined standards, while others may have to undergo extensive testing, risk assessment, and documentation.
CE Marking Consultants in Dubai have expertise in the various directives and regulations governing CE marking, including the Low Voltage Directive (LVD), the Machinery Directive, and the Medical Devices Regulation (MDR), among others.
Manufacturers often find themselves lost in regulatory details and absolute requirements for the products they manufacture.
Risk Assessment and Evaluation of the Product
A key component of the CE marking process is verifying that products comply with safety standards. This is usually done by performing a risk assessment to diagnose potential hazards with the product. A risk assessment is a formal examination of the aspects of the product’s design, function, and use that could be potential risks to the end user or environmental hazards.
Consultants for CE Marking Certification in UAE, assist the manufacturers in assessing potential risks in the early stages of development. By assessing and identifying risks ahead of time, consultants ensure manufacturers have proper safety measures in place to mitigate or entirely remove hazards.
This prevention allows manufacturers to have both time and money saved on safety hazards that are avoided before a product makes it through development and onto the market.
Preparing and Reviewing Technical Documentation
CES marking certification demands proper technical documentation to be built. This documentary evidence proves compliance with these regulatory bodies.
It usually contains design specifications, risk assessments, testing reports, user manuals, and other related documentation. The technical documentation must also be available for regulatory authorities to inspect.
However, for most manufacturers, compiling and organizing technical documentation can be challenging. This involves carrying out specific procedures and providing mandatory documents as outlined by specific directives, which is why CE Marking Consultants in Abu Dhabi will ensure that all the right documentation is prepared, organized, and compliant with specific directives.
They can also assist businesses in spotting incomplete documentation or gaps that could hinder certification.
Product Testing and Evaluation
Some products need to be tested to ensure compliance with safety, environmental, and performance standards to meet CE marking requirements. Testing requirements vary by product type. For instance, a medical device probably will require some clinical evaluation, while an electrical appliance will need an electrical safety evaluation.
Many manufacturers lack the basic knowledge when it comes to the requirements for CE marking, which is where the certification consultants come into play and can help manufacturers oversee which tests their products require. They also help to identify accredited testing laboratories that can perform the required tests.
This early feedback helps manufacturers identify potential safety issues before they become costly recalls or product failures, all while products are still in development and any necessary adjustments can be made in a timely fashion.
Quality Management System Implementation
Part of the CE marking process is confirmation that a product is consistently manufactured to a set standard. For many products, this means that a Quality Management System (QMS) is needed to prove to the manufacturer that they will be able to stay in compliance. This is especially important in medical devices machinery and other products with strict safety regulations associated with them.
Certification consultants can help businesses set or improve their QMS to ensure they meet the requirements. Implementing a strong quality management system from the beginning reduces the chance of non-compliance and product failure.
And while taking an FDA-certified QMS helps with CE marking compliance, a well-implemented QMS also helps businesses manage the quality and safety of products in the long run.
Navigating Conformity Assessment Procedures
Conformity assessment is the procedure used by a manufacturer to prove that the product conforms to the applicable regulation. Depending on the product, a self-certification procedure, a third party notified body assessment, or a combination of both might be required. It is essential to note that the conformity assessment process is not straight from the same path, as the same may vary for different categories of products.
To assist manufacturers in this process, CE marking consultants advise them on which conformity assessment procedure needs to be followed. They assure that the requirement appropriate to the assessment procedure has been fulfilled and that associated documentation and test results supplied by the manufacturer are in accordance.
Addressing Compliance Problems Early On
Oftentimes, as manufacturers are pursuing to get certified, they may run into a hurdle or roadblock during the certification as the team may not have a good understanding of the regulations and not recognize potential issues ahead of time.
CE consultants can identify issues that may lead to a compliance gap before those get too bad. They proactively prepare a roadmap to clarify any deficiencies in documentation, testing, or risk assessment that could ultimately block certification.
By nipping these issues in the bud, consultants save manufacturers from expensive delays, rework, or design changes that could lead to increased costs.
The Final Say!!
The certification is the reflection of the meaning of CE marking. But the regulatory landscape is complex, and navigating it can be a challenge.
CE Marking Certification Consultants in UAE are these experts who ensure businesses can detect a nominal issue sooner, either with regulatory standards, product-related, documentation, or other testing concerns.
In the end, it’s the assurance of compliance with regulatory standards that certification consultants provide to businesses that help not only with compliance but also give customers and regulators confidence in their products and processes.
0 notes
Text
severance essay excerpt under the cut. I think I'm working on a new style? this is mostly close reading, later drafts will bring in outside texts.
and yes half of this is one single footnote. you have met me, yes? also it's house themed bc of course it is (kidding, but it IS originally intended for the monster house thesis sequel). this is from a section titled GOD'S HOUSE, it's about reading irving as a man of faith:
In the land of Lumon, Kier is God; his words are literally pasted into a Bible, as one can see if they pause while Irving flips through the compliance handbook before smushing a deviled egg in it. ("What's For Dinner?") It takes him a while to get to that point, however; in fact, Kier's most loyal devote in the MDR department is Irving, who cannot confidently choose a favorite Virtue—sorry, Principle—because they're all his favorite. It's his fun fact about himself (1) during the icebreaker game they play for Helly's first day.
Which explains why he reacts the way he does to the Perpetuity Wing. If you could step into a perfect replica of God's house, how would you feel? To see the bed where he slept (or a facsimile of it), to eat where he ate (during the coveted Waffle Party), to walk down the halls the way he might have as he wrote the words that propel you day by day? It's not God's house, but it is very, very close—as close as a crack team of corporate archivists can create—and it's just down some winding hallways. What do you do with that knowledge? Do you even find it strange, if you've lived your three years of existence on the same office floor?
And your coworkers are playing sarcastic trivia bingo about this, by the way.
—————
1. In this world, fun facts about oneself are a valuable and intimate currency, often used as incentives (which is to say, bribery) or placating ammunition in the Wellness department, where troubled and troublesome (but not outright disobedient) innies are sent. "These facts are not to be shared outside this room," Ms Casey (a part time innie whose situation eventually is revealed to be dire as well). She binds them in both time and space: "But for now, they’re yours to enjoy." ("Half Loop") On the severed floor, trivia about themselves are used almost like abusive parenting techniques: the Wellness director holds them out, precious gems of self-knowledge, and if the innie reacts in any way to the revelation (positive or negative), the treat is pulled away, not unlike the "blanket training" and other negative reinforcement techniques used to shape obedient children. (More on infantilization in the "Play House" section.) "Please try to enjoy each fact equally, and not show preference for any over the others. That’s ten points off. You have 90 points remaining," Ms Casey chastises flatly later. She adds also, "Please don’t speak further, or all remaining points will be deducted and the wellness session will end." ("Half Loop") Obedience and neutrality is the intended result, even in the guise of new age corporate "wellness" sessions. The carrot is retracted. The session ends, and its font of knowledge is stoppered and locked back behind the door.
#it really is a day for bingo huh#wip tag#do i still have that????? who fucking knows#fic writer tag#not quite but close enough for fannish purposes#txt
1 note
·
View note
Text
CyberProof: Advanced Managed Detection & Response Services
In today’s rapidly evolving cyber threat landscape, organizations face relentless challenges in safeguarding their digital assets. CyberProof stands as a leader in managed detection and response (MDR) services, offering innovative solutions to protect businesses against sophisticated cyberattacks.
What is CyberProof?
CyberProof is a cutting-edge cybersecurity provider specializing in advanced MDR services. With a focus on proactive threat detection, swift response, and continuous innovation, CyberProof helps organizations mitigate risks, ensure compliance, and enhance their overall security posture.
Key Features of CyberProof’s MDR Services
1. Real-Time Threat Detection
CyberProof leverages advanced technologies such as AI, machine learning, and behavioral analytics to identify threats in real time. Their 24/7 monitoring ensures potential risks are detected and mitigated before they escalate.
2. Proactive Incident Response
With a dedicated incident response team, CyberProof ensures rapid containment and eradication of threats. Their response protocols are designed to minimize downtime and prevent data loss during a breach.
3. Comprehensive Threat Intelligence
By harnessing global threat intelligence and contextual insights, CyberProof stays ahead of emerging cyber threats. This enables organizations to prepare for and counteract advanced attack techniques.
4. Integration with Existing Systems
CyberProof’s services seamlessly integrate with an organization’s existing security infrastructure, ensuring cost efficiency and operational continuity.
5. Customized Security Solutions
Every organization’s security needs are unique. CyberProof tailors its services to align with business goals, risk appetite, and compliance requirements.
How CyberProof Stands Out in MDR Services
Advanced Automation and AI
CyberProof employs advanced automation to streamline detection and response processes. By reducing manual intervention, the platform accelerates response times and eliminates redundancies.
Expert Security Analysts
Behind the technology are skilled analysts who bring deep expertise in cybersecurity. Their collaboration with automation tools ensures accurate threat detection and mitigation.
Focus on Business Outcomes
CyberProof emphasizes aligning cybersecurity strategies with business objectives, ensuring that their services drive measurable value.
Industries Served by CyberProof
CyberProof serves a wide range of industries, including:
Finance: Protecting sensitive financial data and ensuring compliance with regulatory standards.
Healthcare: Safeguarding patient information and mitigating risks to critical systems.
Retail: Securing customer data and preventing fraud in an increasingly digital marketplace.
Manufacturing: Addressing vulnerabilities in industrial control systems and IoT devices.
Benefits of Choosing CyberProof
Reduced Risk: Effective threat management minimizes the risk of breaches and financial losses.
Cost Efficiency: Automation and integration optimize resource usage, reducing operational costs.
Scalability: Flexible solutions grow with your organization’s needs.
Regulatory Compliance: Ensure adherence to industry standards such as GDPR, HIPAA, and PCI DSS.
Future-Proof Your Cybersecurity with CyberProof
As cyber threats evolve, the need for advanced MDR services like those offered by CyberProof has never been greater. By combining cutting-edge technology with expert insights, CyberProof helps organizations stay one step ahead of adversaries while maintaining focus on their core business operations.
1 note
·
View note
Link
A new report by MedTech Europe has revealed that the struggle for manufacturers to meet the requirements of the EU’s MDR and IVDR is ongoing. The post New report reveals ongoing industry challenges around EU’s IVDR and MDR compliance appeared first #BioTech #science
0 notes
Text
#Medical Devices#Regulatory Compliance#Product Labeling#FDA Regulations#UDI (Unique Device Identification)#MDR (Medical Device Regulation)#Labeling Standards#Risk Management#Health Canada Regulations#Clinical Labeling#Device Instructions for Use#Packaging and Labeling#Medical Device Safety
0 notes
Text
Medical Device Regulatory Recap 2024 by OMC Medical Limited

In 2024, OMC Medical Limited achieved remarkable milestones while adapting to significant regulatory developments, reinforcing its global presence.
Regulatory Recap 2024
Key Changes That Impacted Business
Compliance with evolving EU MDR and FDA requirements, especially for digital health and Software as a Medical Device (SaMD).
Introduction of region-specific regulations in Asia and the Middle East.
Gaining Valuable Insights for Future Compliance
Improved preparedness for global regulatory shifts.
Enhanced processes for efficient market approvals.
Achievements
Recognized in the Historic Record of NHS 75 book, published by the Houses of Parliament.
Expanded with offices in two major markets, totaling seven global locations.
This year solidified OMC’s position as a leader in medical device regulatory solutions.
Read below to get more detailed information.
Regulatory News Recap 2024
0 notes
Text
youtube
Summary
🔒 Cybersecurity Challenges for SMBs: Small to medium-sized businesses (SMBs) face significant cyber threats but often lack the resources for robust in-house security.
🛡️ Benefits of MDR and SOC Services:
Specialized Expertise: Provides access to experienced security professionals.
24/7 Monitoring: Ensures real-time threat detection and response.
Cost-Effective: Avoids large expenses on in-house resources.
Regulatory Compliance: Helps meet requirements like HIPAA and GDPR.
Enhanced Productivity: Allows businesses to focus on core activities.
0 notes