#bayesian phylogeny
Explore tagged Tumblr posts
Visit Tumblr Blog
Explore Tumblr blogs with no restrictions, modern design and the best experience.
Last Seen Tumblr Blogs





Fun Fact
The total number of visits Tumblr.com received during January 2021 is 327 million.
Text
Idk who "mrbeast" is in the slightest all I know is every time someone brings him up I briefly go "wait why are people talking about a bayesian inference phylogenetics program on tumblr" before I remember that it's mrBAYES, and that's not what people are talking about

#bayesian phylogeny#bayes#mrbayes#mrbeast#loving my “Idk I'm an adult” era of ignorance rn#phylogenetics
154 notes
·
View notes
Text
"An interesting article Language trees with sampled ancestors support a hybrid model for the origin of Indo-European languages in one of the most highly cited and leading scientific journals Science, which was published yesterday!
It is an excellent example of modern humanitarian research on the origin of Indo-European languages. Bayesian analysis, probabilistic models, language phylogeny (!), involvement of paleogenetic data - and we have the most accurate answer to the question about which dissertations on philology were defended and spears were broken for two centuries.
Indo-European languages turned out to be much older than previously thought - ~6120 years BC!"
How convenient for them me not having money to pay for the full article because:


What the hell.
Priscus of Panium with Excerpta de Legationibus and the first mentioned Ukrainian words goes f*ck himself, as I understand correctly.
Grand Dutchy of Lithuania documents go f*ck themselves too in this case.
6 notes
·
View notes
Text
Fwd: Course: Online.InferenceWithBEAST2.Nov18-29
Begin forwarded message: > From: [email protected] > Subject: Course: Online.InferenceWithBEAST2.Nov18-29 > Date: 13 November 2024 at 05:28:53 GMT > To: [email protected] > > > Dear colleagues, > > There are a few slots available for the online course "Bayesian > phylogenetic inference with BEAST2". > > Online live sessions on November 18th, 20th, 22nd, 25th, 27th, and 29th, > 2024, from 15:00 to 18:30 (Madrid time zone) > > Instructors: Dr. Jo�lle Barido-Sottani [1] (Ecole Normale Sup�rieure de > Paris, France) and Dr. Bethany Allen [2] (ETH Zurich, Switzerland) > > Course Overview: > > Bayesian phylogenetic inference is a powerful tool for reconstructing > phylogenies while accounting for complex evolutionary dynamics. It > allows prior knowledge to be integrated into the inference, and also > provides a detailed picture of the uncertainty present in the dataset. > However, the number and complexity of the available models and options > can be daunting for users and can make it difficult to apply inference > tools effectively in practice. > In this workshop, participants will learn the theoretical concepts > underlying the different models involved in Bayesian phylogenetic > inference, and get hands-on experience using these models in BEAST2. > Particular attention will be given to more complex tree models, such as > the fossilized birth-death model used to integrate past information into > phylogenies, as well as rate-heterogeneous models which allow for > variations in evolutionary dynamics across clades. Finally, the course > will give practical information on setting up and troubleshooting > analyses in BEAST2. > > Registration and more information: > https://ift.tt/jx0XyTw > > Best wishes > > Sole > > > Soledad De Esteban-Trivigno, PhD > Director > Transmitting Science > https://ift.tt/y1fJVbc > > Twitter @SoleDeEsteban > Orcid: https://ift.tt/TcWXyRI > > Under the provisions of current regulations on the protection of > personal data, Regulation (EU) 2016/679 of 27 April 2016 (GDPR), we > inform you that personal data and email address, collected from the data > subject will be used by TRANSMITTING SCIENCE SL to manage communications > through email and properly manage the professional relationship with > you. The data are obtained based on a contractual relationship or the > legitimate interest of the Responsible, likewise the data will be kept > as long as there is a mutual interest for it. The data will not be > communicated to third parties, except for legal obligations. We inform > you that you can request detailed information on the processing as well > as exercise your rights of access, rectification, portability and > deletion of your data and those of limitation and opposition to its > treatment by contacting Calle Gardenia, 2 Urb. Can Claramunt de Piera > CP: 08784 (Barcelona) or sending an email to > [email protected] or > https://ift.tt/kZEuK5C. If you consider that > the processing does not comply with current legislation, you can > complain with the supervisory authority at www. aepd.es . > Confidentiality. - The content of this communication, as well as that of > all the attached documentation, is confidential and is addressed to the > addressee. If you are not the recipient, we request that you indicate > this to us and do not communicate its contents to third parties, > proceeding to its destruction. > Disclaimer of liability. - The sending of this communication does not > imply any obligation on the part of the sender to control the absence of > viruses, worms, Trojan horses and/or any other harmful computer program, > and it corresponds to the recipient to have the necessary hardware and > software tools to guarantee both the security of its information system > and the detection and elimination of harmful computer programs. > TRANSMITTING SCIENCE SL shall not be liable. > > Links: > > [1] > https://ift.tt/Zkvdn8r > [2] https://ift.tt/Kjlur1i > > Soledad De Esteban-Trivigno
0 notes
Text
Insects, Vol. 15, Pages 371: A New Species of Scymnus (Coleoptera, Coccinellidae) from Pakistan with Mitochondrial Genome and Its Phylogenetic Implications
In this study, a new species of the subgenus Pullus belonging to the Scymnus genus from Pakistan, Scymnus (Pullus) cardi sp. nov., was described and illustrated, with information on its distribution, host plants, and prey. Additionally, the completed mitochondrial genome (mitogenome) of the new species using high-throughput sequencing technology was obtained. The genome contains the typical 37 genes (13 protein-coding genes, two ribosomal #RNAs, and 22 transfer #RNAs) and a non-coding control region, and is arranged in the same order as that of the putative ancestor of beetles. The AT content of the mitogenome is approximately 85.1%, with AT skew and GC skew of 0.05 and −0.43, respectively. The calculated values of relative synonymous codon usage (RSCU) determine that the codon UUA (L) has the highest frequency. Furthermore, we explored the phylogenetic relationship among 59 representatives of the Coccinellidae using Bayesian inference and maximum likelihood methods, the results of which strongly support the monophyly of Coccinellinae. The phylogenetic results positioned Scymnus (Pullus) cardi in a well-supported clade with Scymnus (Pullus) loewii and Scymnus (Pullus) rubricaudus within the genus Scymnus and the tribe Scymnini. The mitochondrial sequence of S. (P.) cardi will contribute to the mitochondrial genome database and provide helpful information for the identification and phylogeny of Coccinellidae. https://www.mdpi.com/2075-4450/15/5/371?utm_source=dlvr.it&utm_medium=tumblr
0 notes
Text
Turnersuchus: First of the Sea Crocs
A big find for crocodile finds was revealed two days ago. Turnersuchus hingleyae (Hingley's and Turner's crocodile) is the oldest described and basalmost thalattosuchian described so far and is of great importance to slowly figuring out where thalattosuchians come from.
But lets start with a brief introduction to thalattosuchians. As the name already suggests, thalattosuchians are primarily known to have been marine animals (tho exceptions are known). thalattosuchians can broadly be split into two groups. The teleosauroids, which look somewhat similar to what one might call a normal crocodile, and the metriorhynchoids, which especially in the derived members could aptly be described as crocodile mermaids. Below an example of each, on the left Macrospondylus by Nikolay Zverkov and on the right a generalized metriorhynchid by Gabriel Ugueto.


Thalattosuchians such as these two groups, which are sister clades and not successive lineages (so they co-occured rather than one having evolved from the other), were incredibly successful during the Jurassic, evolving enormous forms such as Plesiosuchus, Dakosaurus and Machimosaurus. Their wild success held on throughout the Jurassic until they eventually went extinct in the early Cretaceous. But despite how common and whidespread they are, we don't actually have much of a clue where they come from. Thalattosuchians just kinda appear during the Toarcian and are already found across multiple continents with both groups established. To complicate matters, their position among crocodiles is also rather shaky. Three main hypothesis exist. One is that they are a sister group to crocodyliforms (Protosuchians, Notosuchians and Neosuchians), that they are basal mesoeucrocodylians or that they are Neosuchians related to Pholidosaurids (like Sarcosuchus) and Dyrosaurs.
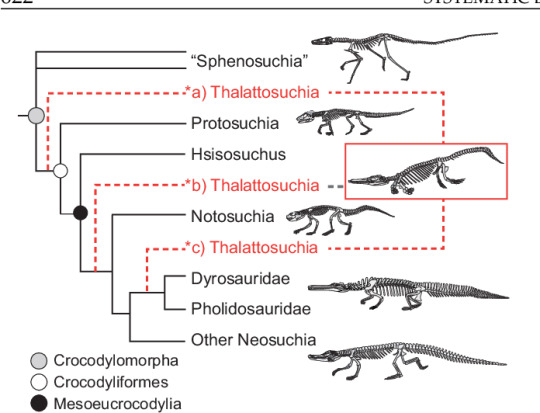
This is where Turnersuchus comes in. Discovered in the Charmouth Mudstone Formation of Dorset, England, this genus is known from the skeletal material belonging to the back of the head, mandible, parts of the forearms and shoulder girdle as well as neck, body and tail vertebrae all preserved in five blocks and a few isolated pieces of bone. From that we can already see general similarities to derived thalattosuchians and basic traits like narrow jaws and reduced forelimbs (tho not nearly as extreme as in metriorhynchids).

The first significant part about this discovery is its age. Turnersuchus is from the Pliensbachian stage of the Jurassic, so it predates any previously diagnostic thalattosuchians. Secondly is its position. As I said above, thalattosuchians are divided into teleosauroids and metriorhynchoids. But Turnersuchus is neither, with both phylogenetic analysis finding it to fall outside of these groups. Now in fairness this is not rock solid, as there is only a single trait excluding it from the derived groups in either analysis, so future works might shake things up. But as things are right now, it's the oldest named and basalmost member of the entire clade. On a sidenote at least the oldest part is bound to change, as the paper mentions a Moroccon teleosauroid currently in press that is even older.
All of this allows for two things. For one, by comparing Turnersuchus with the basal members of both teleosauroids and metriorhynchoids scientists were able to gather a list of traits that appear to be ancestral to the group. In addition, Turnersuchus also preserves some features that separate it from all other thalattosuchians that are also indicative of being an early member. Just as one example a specific part of the basioccipital thats associated with long skulls is poorly developed, which means that while slender the jaws weren't as long as in some later thalattosuchians. A Bayesian analysis was also conducted in an attempt to nail down when thalattosuchians evolved. Now depending on which phylogeny is used (one with thalattosuchians as non-crocodyliforms and another with them as mesoeucrocodylians) you get different times. The former would place their origin in the Norian stage of the Triassic, the later in the Sinemurian stage of the Jurassic. In light of the Moroccon material alluded to by the paper, it would appear that they likely split from other crocodylomorphs sometime in the late Triassic.
And finally to wrap this up let me share the press release artwork to finally give a face to all this information. Now if you've read my post about fossil crocs of 2022 you might already recognize the artist, as she's been on a real streak with illustrating fossil crocs. If you don't know her, I highly recommend checking out her work. I'm of course talking about Júlia d'Oliveira.

And yes, of course I got the Wikipedia page for it ready. Tho with work getting in the way when it was published I almost feared I'd be unable to get to it first.
#turnersuchus#thalattosuchia#metriorhynchidae#teleosauridae#jurassic#paleontology#palaeblr#crocodylomorph#croc#pseudosuchian#marine reptile#long post#Wikipedia#paleontology news#crocodile
117 notes
·
View notes
Photo

Tetrapodophis amplectus is not a snake: re-assessment of the osteology, phylogeny and functional morphology of an Early Cretaceous dolichosaurid lizard
Caldwell et al.
Abstract
The origin of snakes remains one of the most contentious evolutionary transitions in vertebrate evolution. The discovery of snake fossils with well-formed hind limbs provided new insights into the phylogenetic and ecological origin of snakes.
In 2015, a fossil from the Early Cretaceous Crato Formation of Brazil was described as the first known snake with fore- and hind limbs (Tetrapodophis amplectus), and was proposed to be fossorial, to exhibit large gape feeding adaptations (macrostomy) and to possess morphologies suggesting constriction behaviours.
First-hand examination of T. amplectus, including its undescribed counterpart, provides new evidence refuting it as a snake.
We find: a long rostrum; straight mandible; teeth not hooked zygosphenes/zygantra absent; neural arch and spines present and tall with apical epiphyses; rib heads not tubercular; synapophyses simple; and lymphapophyses absent. Claimed traits not preserved include: braincase/descensus parietalis; ‘L’-shaped nasals; intramandibular joint; replacement tooth crowns; haemal keels; tracheal rings; and large ventral scales.
New observations include: elongate retroarticular process; apex of splenial terminating below posterior extent of tooth row; >10 cervicals with hypapophyses and articulating intercentra; haemapophyses with articulating arches; reduced articular surfaces on appendicular elements; rows of small body scales; and reduced mesopodial ossification. The axial skeleton is uniquely elongate and the tail with >100 vertebrae is not short as previously claimed, although overall the animal is small (∼195 mm total length). We assessed the relationships of Tetrapodophis using a revised version of the original morphological dataset, an independent morphological dataset, and these two datasets combined with molecular data. All four were analysed under parsimony and Bayesian inference and unambiguously recover Tetrapodophis as a dolichosaur.
We find that Tetrapodophis shows aquatic adaptations and there is no evidence to support constricting behaviour or macrostomy.
Read the paper here:
https://www.tandfonline.com/doi/full/10.1080/14772019.2021.1983044
25 notes
·
View notes
Text
Move over, Neanderthals. This could actually be our closest human relative
https://sciencespies.com/humans/move-over-neanderthals-this-could-actually-be-our-closest-human-relative/
Move over, Neanderthals. This could actually be our closest human relative
In 1933 a mysterious fossil skull was discovered near Harbin City in the Heilongjiang province of north-eastern China. Despite being nearly perfectly preserved – with square eye sockets, thick brow ridges and large teeth – nobody could work out exactly what it was.
The skull is much bigger than that of Homo sapiens and other human species – and its brain size is similar to that of our own species. Historical events left it without a secure place of origin or date, until today.
Now a team of Chinese, Australian and British researchers has finally solved the puzzle – the skull represents a previously unknown extinct human species. The research, published as three studies in the journal Innovation, suggests this is our closest relative in the human family tree.
Dubbed Homo longi, which can be translated as “dragon river”, it is named after the province in which it was found. The identification of the skull, thought to have come from a 50-year-old male, was partly based on chemical analysis of sediments trapped inside it.
This confirmed it comes from the upper part of the Huangshan rock formation near Harbin City. The formation was reliably dated to the Middle Pleistocene – 125,000 to 800,000 years ago. Uranium series dating, which involves using the known rate of decay of radioactive uranium atoms in a sample to work out its age, showed that the fossil itself is at least 146,000 years old.
Homo longi can now takes its place among an ever increasing number of hominin species across Africa, Europe and Asia.
Fossil human skulls H. erectus (left) and H. longi (furthest right). (Kai Geng)
Constructing a family tree
Determining the historical relationship between fossil species, however, remains one of the most difficult tasks in the study of human evolution.
In recent years, the analysis of ancient DNA has transformed our understanding of the relationship between early populations of modern humans. It has also highlighted how we are different – and similar – to our most immediate relatives, the Neanderthals.
Surviving DNA, however, is very rare for fossil hominins from the Middle Pleistocene, as it tends to degrade over time. Evolutionary relationships must therefore be determined using other evidence. This is usually data on the shape – morphology – of fossils, their age and geographical location.
The Harbin team generated a family tree (“phylogeny”) of human lineages to work out how the species relates to modern humans. This tree is based on morphological data from 95 largely complete fossil specimens of different hominin species living during the Middle Pleistocene, including Homo erectus, Homo neanderthalensis, Homo heidelbergensis and Homo sapiens along with their known ages.
The tree also suggests that five previously unidentified fossils from northeastern China are from Homo longi.
Human family tree. (Ni et al. Innovation, 2021)
It predicts that the common ancestor of Homo longi and Homo sapiens lived approximately 950,000 years ago. Furthermore, it suggests that both species shared a common ancestor with Neanderthals a bit more than 1 million years ago, meaning we may have split from Neanderthals 400,000 earlier than previously thought (we used to think it was 600,000 years ago).
Until now, the Neanderthals were considered our closest relative (according to the study, we split from Homo heidelbergensis some 1.3 million years ago). Debates about the evolution of modern humans and what it is that makes us “human” therefore relied heavily on comparisons to Neanderthals.
But the new discovery pushes Neanderthals one step further away from ourselves and makes simple comparisons between two species much less important to understanding what ultimately makes us who we are.
There are, however, still significant points of concern about the dating of this phylogenetic model, as recognized by the authors. The predicted dates for the common ancestors between human lineages do not match the dates of actual discovered fossils, or those predicted by the analysis of DNA.
For example, this study proposes that there was Homo sapiens in Eurasia at about 400,000 years ago. But the oldest fossil for this species known outside Africa is little more than half this age.
At the same time, the split between Homo sapiens and Neanderthals predicted here at more than 1 million years old does not match the prediction of nuclear DNA analysis, which suggests it happened much later. However, it can be backed up by doing DNA analysis with genetic material taken from the cell’s engine, called the mitochondria.
The older estimates presented by this study may result from the use of new techniques, called Bayesian tip dating, which aren’t normally used in evolutionary studies. These can take into account both morphological and molecular data and make predictions about the possible sequence and date of the divergence of species.
Reconstruction of Homo longi. (Chuang Zhao)
Wider perspective
While the shape of the family tree presented here is likely to stand the test of time, it is still too early to accept these predicted divergence dates as definitive. That said, the research also sheds important light on how human species occurred and spread through the Middle Pleistocene – into all areas of our planet. Crucially, many of these species may have interbreed.
Europe was the origin point for Neanderthals. Meanwhile, the Asian human species Homo erectus was a critical evolutionary step, giving rise to all later hominin species. And now we know that Homo longi evolved in Asia too. It therefore looks like Africa was a destination as well as a point of origin for the spread of human species.
The Harbin cranium also tells another story about human evolution as a science and as an international discipline. Human evolution was originally a European area of interest, focused on evidence from sites in western and central Europe. The discovery of fossils in Africa added great time depth to the origins of the human lineage and led to a common story of the spread of new species out of Africa.
The Harbin cranium reminds us of the vast expanse of Asia, whose fossils and scientists are now coming to the fore. Further insights may come both from the discovery of new species and old figurative art.
In the case of the Harbin cranium, it is the application of new techniques of analysis that has brought old specimens back into active use. Asia is now in the driving seat of the study of human evolution.
Anthony Sinclair, Professor of Archaeological Theory and Method, University of Liverpool.
This article is republished from The Conversation under a Creative Commons license. Read the original article.
#Humans
#06-2021 Science News#2021 Science News#Earth Environment#earth science#Environment and Nature#freaky#Nature Science#News Science Spies#oddities#Our Nature#outrageous acts of science#planetary science#rare#scary#Science#Science Channel#science documentary#Science News#Science Spies#Science Spies News#Space Physics & Nature#Space Science#weird#Humans
0 notes
Text
CHARACTER ANALYSIS
CHARACTER STEP MATRIX
As reviewed earlier, assigning a character state transformation determines the number of steps that may occur when going from one character state to another. Computerized phylogeny reconstruction algorithms available today permit a more precise tabulation of the number of steps occurring between each pair of character states through a character step matrix. The matrix consists of a listing of character states in a top row and left column; intersecting numbers within the matrix indicate the number of steps required, going from states in the left column to states in the top row. For example, the character step matrix of Figure 2.5A illustrates an ordered character state transformation series, such that a single step is required when going from state 0 to state 1 (or state 1 to state 0), two steps are required when going from state 0 to state 2, etc. The character step matrix of Figure 2.5B shows an unordered transformation series, in which a single step is required when going from one state to any other (nonidentical) state. Character step matrices need not be symmetrical; that of Figure 2.5C illustrates an ordered transformation series but one that is irreversible, disallowing a change from a higher state number to a lower state number (e.g., from state 2 to state 1) by requiring a large number of step changes (symbolized by “∞”). Character step matrices are most useful with specialized types of data. For example, the matrix of Figure 2.5D could represent DNA sequence data, where 0 and 1 are the states for the two purines (adenine and guanine) and 2 and 3 are the states for the two pyrimidines (cytosine and thymine; see Chapter 14). Note that in this matrix the change from one purine to another purine or one pyrimidine to another pyrimidine (each of these known as a “transition”) requires only one step, being biochemically more probable to occur, whereas a change from a purine to a pyrimidine or from a pyrimidine to a purine (termed a “transversion”) is given five steps, being more biochemically less likely. Thus, in a cladistic analysis, the latter change will be given substantially more weight.
DNA sequence data may be transformed in a more complicated evolutionary model, based on a number of parameters, such as branch length, codon position, base frequency, or transition/transversion ratio. Such models of evolution are an integral component of maximum likelihood and Bayesian analyses (see later discussion).
Chapter Two Plant Systematics Second Edition Michael G. Simpson
1 note
·
View note
Note
What software do you use to make cladograms?
That depends on which cladograms you’re asking about. For something quick and simple like the one below, I use a specialized software called TreeGraph.

For the time-scaled bird phylogenies I posted a while back, I used... Microsoft PowerPoint, believe it or not. (There are also packages in R that can make similar graphics.)

For the Cartoon Guide to Vertebrate Evolution? Hand drawn in GIMP using a drawing tablet (like most of my drawings).

For running actual phylogenetic analyses in my research? TNT (for parsimony) and MrBayes (for Bayesian methods).
11 notes
·
View notes
Text
Saurian DevLog #47
Hey all,
We've been working really hard on getting the next patch out, as well as the art book so all of the team is in ultra-busy mode at the moment. Henry has some information for you on what is to come with regards to the patch and I thought I'd drop some science seeing as an interesting new paper just dropped that we got a first-look at.
Henry
The programming team has been busy working out the last bugs from the gradual ontogeny system implementation, as well as some other longer-standing ones. After some last-minute sprints, we're happy to say it's ready! Patch notes can be found here - please let us know if you encounter any issues. Otherwise, enjoy!
Tom
A bit ago we were contacted by palaeontologist Mike Lee, asking if they could use our Thoracosaurus render in the press release of a new paper he had in the works featuring this animal. Always happy to have our art used for sci-comm, we said yes and Mike was kind enough to send us a pre-print of the paper in question. It turned out to be a pretty cool study, and had some implications for us which I thought would be interesting to discuss. Seeing as it has now been published I thought I'd talk about it here today.
To be clear, it won't effect how the animal will appear in game, but I did have to re-write the profiles for all our crocodylomorphs in the art book and I will have to modify the encyclopedia entries as well.
Thoracosaurus is a genus of large (5m or more) marine crocodylomorph from the late cretaceous of North America (including Hell Creek) and Europe. Since it was first discovered, Thoracosaurus has been considered an early member of the gharial lineage, with many similar aspects of the skeleton indicating this might have been the case. Computational phylogenetic analysis - computer run analysis that hypothesise the relationships of organisms - also consistently came to the same conclusion.
Borealosuchus is another crocodylomorph from Hell Creek that sometimes comes out as close to gharials, and sometimes in other positions close to the base of living crocodilians.
Traditional morphological studies had found gharials to be the earliest branching group of crocodylians. This was consistent with the 72 million year age of the earliest species of Thoracosaurus. however, new relationships and divergent dates received from molecular studies did not, with gharials being a reletively young group (40 Ma) more closely related to crocodiles than alligators. In this scenario, Thoracosaurus appears far before when gharials are supposed to have diverged from crocodiles.
This is where the new study by Mike Lee and his colleague Adam Yates comes in. They found that when running extremely complete phylogenies taking molecular, morphological and fossil age data into account, this disparity was resolved.
The inclusion of data related to age gaps between groups (tip-dating methodology) proved to be the difference, as without these methods the relationships came out similar to previous studies. The inclusion of tip-dating Bayesian approaches found Thoracosaurus and Borealosuchus to not be related to gharials at all, but out side of crocodylia. The position of gharials here agrees with molecular approaches, being young and close to crocodiles. This means than the gharial-like features of Thoracosaurus are all convergently evolved, probably as adaptions for catching fish.
So what does this mean for us, on a surface level? Well probably not a lot in terms of appearance, behaviour and ecology. What it does mean is than both Thoracosaurus and Borealosuchus are no longer crocodylians! They are stem-crocodylians and each other's closest reletives. So our crocodylian count in Saurian has dropped from 3 to 1, with early alligatoroid Brachychampsa being the only one left.
61 notes
·
View notes
Text
Phylogenetics
» motivation
goal: build a tree that gives us the evolutionary history of genomes, beyond simple sequence alignment similarities
before the advent of DNA sequencing, phylogenetic tree-building relied on phenotype
[often, this missed the distinction between true evolutionary similarity and convergent evolution]
why do we need the evolutionary history of organisms?
to trace pathogen evolution and pre-empt/treat pathologies
to measure and maintain biodiversity
to analyze/understand more about the evolution of a specific protein function in biochemistry research
i.e, ancestral sequence reconstruction of closely related specifically-binding proteins that possibly had a nonspecific common ancestor
» phylogenetic trees
rooted trees [centered around a selected LUCA] and unrooted trees, in either vertical or horizontal direction
axis perpendicular has no meaning
axis parallel to the branches may have meaning depending on cladogram (none) | chronogram (time) | phylogram (change/differences/number of mutations between)
dated, resolved bifurcated nodes [branches into 2 lines at every split] or undated, unresolved multifurcated nodes
internal branch and external branch – older and more recent branches
the individual species taxa and the clusters of related species in clades
» phylogeny algorithms
[scoring algorithms] best tree among the ones you've selected
parsimony:
generate unrooted tree models for different mutations
count the number of changes required between different branches
least changes = best tree, assuming mutations are all equally likely
issue w/ OG: not all mutations are equally likely, so in reality changes would have to be weighted
transitions are more likely than transversions due to nucleotide bases' chemical similarities
G's are most easily oxidized, which gives it a higher likelihood of mutating
organisms as a whole have differing baseline nucleotide preferences [GCAT proportions & codon preferences vary, so evolutionary pressures shift in one way or another]
modified/weighted parsimony
use maximum likelihood that makes the given data the most likely: likelihood L(d) = P(Data | H tree + evolutionary model) » L(d) = P(Data | H)
use Bayesian approach based on Bayes formula: P (tree T & evolutionary parameters ø) = [P(T, ø) • P(data D | T, ø)] / P(D)
for Bayes, you don't need to choose models to test, you can simply heuristically sample a few of them » this gives a probability distribution across different T's and ø's
[searching algorithms] selecting trees to be scored in the first place
# of unrooted bifurcating trees available: B(T) = ∏(2n – 5) with n = 3, creating an exponential function
finding ALL the trees is not feasible as the calculation becomes intensive
instead, use a heuristic algorithm to quickly find an alright (but not optimal) solution
bootstrapping to optimize: build a tree from a random subset of sequences with replacement
do this multiple times and note the trees that keep repeatedly being created as the more optimal ones
look at all the trees you've generated and see which % of them form certain branches
(i.e., 100% of trees agreeing on a branch means that the branch likely actually exists!)
phylogeny applications
for a universal Tree of Life™
a nice gene to use: ribosome small subunit that is needed for translation, which has both fast and slow-evolving regions
add in some ribosomal proteins to make the tree more accurate
are eukaryotes a smaller branch of archaea, or is there an equal eukarya/archaea branching with a split off from bacteria?
lateral gene transfer really makes things difficult
tracing epidemics
phylogenetics methods are useful for tracing flu/COVID epidemics & HIV
HIV phylogeny in criminal courses cases » victim & patient strain similarity via multiple scoring & tree-building methods for accuracy
covid-specific phylogeny – pangolin's con_lineages.org that lists all the mutations and keeps track of the COVID tree
0 notes
Text
Fwd: Course: Online.PhylogeneticSpeciesDistribution.May12-14
Begin forwarded message: > From: [email protected] > Subject: Course: Online.PhylogeneticSpeciesDistribution.May12-14 > Date: 4 February 2025 at 05:29:44 GMT > To: [email protected] > > > > ONLINE COURSE – Phylogenetic Species Distribution Modelling using R > (PSDM01) This course will be delivered live > > https://ift.tt/HbqBy7n > > 12th - 14th May2025 > > Instructor-Dr. Morales Castilla Ignacio > > Please feel free to share! > > COURSE OVERVIEW:In this three-day course, we introduce species > distribution models (SDMs) and ways to incorporate phylogenetic > information into single species models using R. We begin by providing > an overview on the use of SDMs as a central tool for ecologists and > evolutionary biologists, review and implement common SDM approaches and > introduce hybrid models,which use the information in functional traits to > complement the models. We then justify the rationale for using phylogenetic > information in absence of functional trait data and show how to incorporate > phylogenetic information in SDMs (day 1). We review examples of practical > implementation of PSDMs to both present and future climate scenarios > (day 2).Finally, we overview more advanced approaches of incorporating > phylogenies into models(the Bayesian Phylogenetic Mixed Model) and how > to project model results into a spatial context (day 3). > > Please email [email protected] with any questions. > > February > > ONLINE COURSE – Machine Vision using Python (MVUP01) This course will > be delivered live > > ONLINE COURSE – Machine Learning using Python (MLUP01) This course > will be delivered live > > ONLINE COURSE – Species Distribution Modelling With Bayesian Statistics > Using R (SDMB06) This course will be delivered live > > ONLINE COURSE – Remote sensing data analysis and coding in R for ecology > (RSDA01) This course will be delivered live > > ONLINE COURSE – Introduction to generalised linear models using R and > Rstudio (IGLM08) This course will be delivered live > > ONLINE COURSE – Community Analytics in Ecology and Evolutionary Biology > for Beginners (CAFB01) This course will be delivered live > > March > > ONLINE COURSE – Introduction To Mixed Models Using R And Rstudio > (IMMR09) This course will be delivered live > > ONLINE COURSE – Stable Isotope Mixing Models using SIBER, SIAR, MixSIAR > (SIMM11) This course will be delivered live > > ONLINE COURSE – Multivariate Analysis Of Ecological Communities Using > R With The VEGAN package (VGNR07) This course will be delivered live > > May > > ONLINE COURSE – Phylogenetic Species Distribution Modelling using R > (PSDM01) This course will be delivered live > > ONLINE COURSE – Movement Ecology Using R(MOVE07) This course will be > delivered live > > June > > ONLINE COURSE – Tidyverse for Ecologists and Evolutionary Biologists > (TIDY01) This course will be delivered live > > July > > ONLINE COURSE – Path analysis, structural equations and causal inference > for biologists (PSCB03) > > October > > ONLINE COURSE – Bioacoustics Data Analysis using R (BIAC05) This course > will be delivered live > > -- > > Oliver Hooker PhD. > > PR stats > > > Oliver Hooker
0 notes
Text
Descargar windows 7 mega 32 bits 無料ダウンロード.Download .NET Framework
Descargar windows 7 mega 32 bits 無料ダウンロード.MEGA - パソコン用

The Latest Version:.ダウンロード Windows 7 Home Premium 32 bits 用 Windows 無料 |
MEGA for Windows and Linux(32 and 64 bit) and macOS is now available. This is a bug fix release and is the current stable release Free for 32 bit win download. Development Tools downloads - MEGA by Tamura K, Peterson D, Peterson N, Stecher G, Nei M, and Kumar S and many more programs are available for instant and free download · Downloads for building and running applications Framework. Get web installer, offline installer, and language pack downloads Framework
Descargar windows 7 mega 32 bits 無料ダウンロード.Download ClocX - freeware analog clock
Free for 32 bit win download. Development Tools downloads - MEGA by Tamura K, Peterson D, Peterson N, Stecher G, Nei M, and Kumar S and many more programs are available for instant and free download · Windows PCにMEGA をダウンロードしてインストールします。 あなたのコンピュータにMEGAをこのポストから無料でダウンロードしてインストールすることができます。PC上でMEGAを使うこの方法は、Windows 7/8 / / 10とすべてのMac OSで動作します。 These are ISO images created with ImgBurn from clean Windows 7 Professional SP1 install disks (32 bit and 64 bit respectively). These ISOs are English by default. Hopefully these are of use to someone who wishes to create virtual machines, or even install on older hardware!User Interaction Count: 73K
Version 11 adds new timing methods and is optimized for working with larger data. Calibration densities, tip dating, and a rate auto-correlation test have been added to MEGA.
The Maximum Likelihood system has been optimized for memory efficiency in version The Tree Explorer toolbar has been updated to be more intuitive and accessible. In the past 25 years, the MEGA software has been downloaded more then 2. Sophisticated and user-friendly software suite for analyzing DNA and protein sequence data from species and populations. MEGA This is a bug fix release and is the current stable release.
MEGA Molecular Evolutionary Genetics Analysis version 11 Version 11 adds new timing methods and is optimized for working with larger data. MEGA 11 introduces expanded relaxed-clock dating methods Calibration densities, tip dating, and a rate auto-correlation test have been added to MEGA. Memory efficiency of the ML system has been improved The Maximum Likelihood system has been optimized for memory efficiency in version Redesigned Tree Explorer toolbar The Tree Explorer toolbar has been updated to be more intuitive and accessible.
MEGA Software Celebrates Silver Anniversary In the past 25 years, the MEGA software has been downloaded more then 2. Instructional Videos Now Available Learn how to use MEGA from video tutorials created by MEGA users. Sequence Analyses Phylogeny Inference Model Selection Dating and Clocks Ancestral States Selection and Tests Sequence Alignment. Statistical Methods Maximum Likelihood Distance Methods Ordinary Least Squares Maximum Parsimony Composite Likelihood Bayesian.
Documentation Online Manual MEGA 1.
0 notes
Text
@whereofonecannotspeak I can answer this! Normally when we're talking about "relatedness" in this context, we're talking about divergence times. Now, there are ways to quantitatively estimate genetic relatedness - if you're curious you can look up fixation index (Fst). Fst compares variation within a population to variation between populations, using the frequency of nucleotides (A, T, C, and G) at shared sites. For example, if my "forest population" of a particular bird has all A's at a certain site, and my "field population" has all T's, the Fst at that site is 1. If both populations have all T's, the Fst is 0. If the forest population has mostly A's and the field population has mostly T's, the Fst is a little less than 1. You can scale this up across many sites (and many populations) in the genome to get an idea of relatedness.
However, this isn't super useful for looking at different species, since it doesn't tell us HOW they are related. There are also statistical issues with Fst as your organisms become more distantly related. Instead, we use phylogenies (essentially family trees):
When we're looking at a phylogeny, we can say that cows (bovids) and whales (cetaceans) are more closely related to each other than to horses, since they shared a more recent common ancestor. You can construct phylogenies by taking sequence data from one species and comparing it to another, via a sequence evolution model. You can use a single conserved gene, a collection of conserved genes, or even genome-wide shared sites, which you align together. Methods to generate phylogenies are getting very sophisticated, with a lot of people using Bayesian methods nowadays.

That rabbit/hare post is messing me up. I’d thought they were synonyms. Their development and social behavior are all different. They can’t even interbreed. They don’t have the same number of chromosomes. Dogs, wolves, jackals, and coyotes can mate with each other and have fertile offspring but rabbits and hares cant even make infertile ones bc they just die in the womb. Wack.
340K notes
·
View notes
Text
Recent Perusals
Citations are in Harvard format:
Asano, M., Khrennikov, A., Ohya, M., Tanaka, Y. and Yamato, I., 2015. Quantum adaptivity in biology: from genetics to cognition. Heidelberg: Springer.
“The aim of this book is to introduce a theoretical/conceptual principle (based on quantum information theory and non-Kolmogorov probability theory) to understand information processing phenomena in biology as a whole — the information biology — a new research field, which is based on the application of open quantum systems (and, more generally, adaptive dynamics [173, 26, 175]) outside of physics as a powerful tool. Thus this book is about information processing performed by biosystems.“ (P. XI)
Radder, H. ed., 2003. The philosophy of scientific experimentation. University of Pittsburgh Press.
“Until the advent of computers, the primary tool physicists had at their disposal for representing their theoretical understanding of the mechanics and dynamics of material systems had been the differential equation, and their principal task was to relate the solutions of these equations to observed experimental effects. But differential equations are notoriously difficult to solve once they depart from the linear domain, and especially so when representing the interactions of many bodies. Thus, prior to the computer, the study of complex, nonlinear phenomena by physicists had been limited [...]” (Chapter 10, P. 200-201)
Blackmore, S., 1999. The meme machine—With a foreword of Richard Dawkins.
“I like to ask a simple question – indeed I shall use this question again in several different contexts. Imagine a world full of hosts for memes (e.g. brains) and far more memes than can possibly find homes. Now ask, which memes are more likely to find a safe home and get passed on again? This is a reasonable way to characterise the real world we live in. Each of us creates or comes across countless memes every day. Most of our thoughts are potentially memes but if they do not get spoken they die out straight away. We produce memes every time we speak, but most of these are quickly snuffed out in their travels.” (P. 37)
Krantz, D., Luce, D., Suppes, P. and Tversky, A., 1971. Foundations of measurement, Vol. I: Additive and polynomial representations.
“Little seems possible in the way of a careful analysis of an attribute until means are devised to say which of two objects or events exhibits more of the attribute. Once we are able to order the objects in an acceptable way, we need to examine them for add itional structure, for example, by selecting two or more factors that affect the ordering. Then begins the search for qualitative laws satisfied by the ordering and the additional structure. In contrast to fundamental physical measurement, which is typically oned imensional (see, however, Chapter I O), many of the theories of measurement that appear applicable to behavioral problems are inherently multidi mensional, and so the measurement theories deal simultaneously with several measures and the laws connecting them.” (P. 32)
Jäger, G. and List, J.M., 2016. Statistical and computational elaborations of the classical comparative method.
“According to the subjective or Bayesian interpretation, the probability of an outcome quantifies the degree of certainty one has about this outcome. If, for instance, an election forecast says that candidate X has a 60% chance of winning the next election, this expresses the forecasters’ degree of certainty on the basis of their knowledge, not some relative frequency. This interpretation seems well-suited for historical reconstruction as well. A statement such as “With 60% probability, Italic and Celtic form a common sub-group of Indo-European.” is coherent unter the Bayesian, but not under the frequentist interpretation.“ (P. 26)
Kita, E., 2011 Evolutionary algorithms. Rijeka: InTech.
“Evolutionary algorithms (EAs) are the population-based metaheuristic optimization algorithms. Candidate solutions to the optimization problem are defined as individuals in a population, and evolution of the population leads to finding better solutions. The fitness of individuals to the environment is estimated and some mechanisms inspired by biological evolution are applied to evolution of the population. Genetic algorithm (GA), Evolution strategy (ES), Genetic programming (GP), and Evolutionary programming (EP) are very popular Evolutionary algorithms.” (P. ix)
Hall, D. and Klein, D., 2010, July. Finding cognate groups using phylogenies. In Proceedings of the 48th Annual Meeting of the Association for Computational Linguistics (pp. 1030-1039). Association for Computational Linguistics.
“In this paper, we present a new generative model for the automatic induction of cognate groups given only (1) a known family tree of languages and (2) word lists from those languages. A prior on word survival generates a number of cognate groups and decides which groups are attested in each modern language. An evolutionary model captures how each word is generated from its parent word. Finally, an alignment model maps the flat word lists to cognate groups. Inference requires a combination of message-passing in the evolutionary model and iterative bipartite graph matching in the alignment model.” (P. 1031)
#reading music: Dreamrider by Lazerhawk#I really value free reading time. good stuff#that first one is super interesting#linguistics#reading#science!
1 note
·
View note
Text
Practical Speedup of Bayesian Inference of Species Phylogenies by Restricting the Space of Gene Trees
MBE latest: http://dlvr.it/RQQlJK
0 notes