#rokotteet
Explore tagged Tumblr posts
Visit Tumblr Blog
Explore Tumblr blogs with no restrictions, modern design and the best experience.
Last Seen Tumblr Blogs





Fun Fact
Tumblr has been providing a Korean-language service since 2013.
Text
Pfizer/BioNTech C4591001 Koetutkimus - Tarkastuskertomus - v1 (2024-05-31)
Pfizer/BioNTech C4591001 Koetutkimus - Tarkastuskertomus - v1 (2024-05-31)
Tiivistelmä
Tässä katsauksessa pyritään käsittelemään Pfizer/BioNTech C4591001 -tutkimuksen tietojen tarkastelussa esiin tulleita merkittäviä poikkeamia ja ristiriitaisuuksia, joilla voi olla syvällisiä vaikutuksia yleisön luottamukseen ja sääntelynormeihin, jos niitä ei tutkita asianmukaisesti ja avoimesti.
Ilmiantajatodistukset
Ventavia Research Groupin entinen aluejohtaja Brook Jackson raportoi järjestelmällisistä ongelmista, kuten asianmukaisen tietoon perustuvan suostumuksen puuttumisesta, kelvottomien osallistujien rekisteröinnistä, tietojen väärentämisestä ja säännösten noudattamatta jättämisestä. Huolimatta siitä, että FDA:lle ilmoitettiin näistä huolenaiheista, niihin ei puututtu asianmukaisesti, eikä kriittisiä toimipaikkoja tarkastettu, mikä herättää kysymyksiä viranomaisvalvonnasta.
Augusto Roux’n tapaus
Tohtori Augusto Roux, joka osallistui tutkimukseen Argentiinassa, koki vakavia haittavaikutuksia rokotteen saamisen jälkeen, mukaan lukien sydänpussitulehdus, joka alun perin luokiteltiin sponsorin pyynnöstä virheellisesti mahdolliseksi COVID-19-tapaukseksi. Hänen haittavaikutusilmoituksensa oli puutteellisesti dokumentoitu, ja hänen oireitaan yritettiin luokitella uudelleen. Roux’n tapaus osoittaa, että yleisiä terveyssääntöjä on rikottu vakavasti, ja Argentiinassa on meneillään rikostutkinta hänen todistamistaan tapahtumista.
Satunnaistamisluvuissa esiintyvät poikkeamat
Kahden tutkijan 26. marraskuuta 2020 ja 29. maaliskuuta 2021 tuottamien tiedostojen analyysi paljasti selittämättömiä ristiriitaisuuksia satunnaistettujen koehenkilöiden määrässä. Esimerkiksi Argentiinassa sijaitsevassa tutkimuskohteessa 1231/4444 havaittiin vähenemistä 5776 koehenkilöstä 5615 koehenkilöön, mikä tarkoittaa 161 koehenkilön ”satunnaistamiskatoa”, mitä ei voida selittää koehenkilöiden siirtymisellä tutkimuskohteiden välillä tai alle 16-vuotiaiden koehenkilöiden rekrytoinnilla. Kaiken kaikkiaan 1203 koehenkilöä ”katosi satunnaistamisessa” 108:ssa 153:sta tutkimuspaikasta, mikä kertoo merkittävästä poikkeavuudesta.
Puuttuvat kohdetunnisteet
Tutkimushenkilöiden tunnisteissa havaittiin poikkeavuuksia, ja 301 puuttuvan tunnisteen määrä vaikutti suhteettomasti tiettyihin tutkimuspaikkoihin, erityisesti Argentiinassa. Tämän ongelman tutkiminen viittaa siihen, että tietojen käsittelyssä ei ole kyse satunnaisuudesta, mikä voisi viitata siihen, että koehenkilöiden tietoja on poistettu tarkoituksellisesti.
Kuolemantapausten ilmoittaminen myöhässä
Michels et al. ja Jeyanthi Kunadhasanin Australian Therapeutic Goods Administration (TGA) kanssa käymässä kirjeenvaihdossa korostetaan kuolemantapausten viivästyneen ilmoittamisen merkitystä EUA-luvuissa (Emergency Use Authorization). New England Journal of Medicine -lehdessä julkaistussa Polackin ym. tutkimuksessa raportoitiin kuudesta kuolemantapauksesta, kun taas Pfizerin sisäisissä tiedoissa oli jo kahdeksan kuolemantapausta, joista kahta BNT162b2-haaran kuolemantapausta ei julkistettu. Tällaiset viivästykset ja julkistamatta jättämiset, erityisesti sydäntapahtumiin johtaneiden kuolemantapausten osalta, herättävät vakavia huolenaiheita avoimuudesta ja mahdollisista yleisten toimintaohjeiden rikkomisista.
Protokollapoikkeamiin liittyvät sääntöjenvastaisuudet
Ilman sokkotarkastelua kirjatut protokollapoikkeamat, jotka ovat vastoin tavanomaisia kliinisten tutkimusten käytäntöjä, osoittivat merkittäviä epäsuhtaisuuksia hoitoryhmissä. Merkittäviä poikkeamia olivat muun muassa tutkimussuunnitelmassa määriteltyjen toimenpiteiden epäasianmukainen suorittaminen, virtsan raskaustestien tekemättä jättäminen ja muiden tutkimukseen kuulumattomien koronavirusrokotteiden saaminen, mikä viittaa siihen, että osallistujia itseään ei oltu sokkoutettu, ja tutkimuspaikan henkilökuntaa kohdeltiin lisäksi epäasianmukaisesti.
Ilmoittamattomat haittatapahtumat
Havaittiin tapauksia, joissa haittavaikutuksista ilmoitettiin liian vähän, kuten tapaus, jossa ”rintakipua” ei kirjattu haittavaikutuslokiin, vaan se kirjattiin vasta tapausraporttilomakkeisiin myöhemmän vakavan haittatapahtuman (SAE) vuoksi. Toinen esimerkki koski vakavan sydänpussitulehduksen virheellistä luokittelua COVID-19-sairaudeksi, mikä osoitti hyvien kliinisten käytäntöjen rikkomisen.
Haittavaikutusten uudelleenkelpuutus
Augusto Roux’n todistus paljastaa, että protokollan heikkouksia hyväksikäytettiin järjestelmällisesti, mikä johtaa siihen, että haittavaikutukset luokitellaan laajalti uudelleen COVID-19-oireiksi. AESPID-tietojen analyysi osoittaa, että ainakin 1209 haittavaikutusta 767 koehenkilön kohdalla luokiteltiin uudelleen vaikuttaen teho-, turvallisuus- ja immunogeenisuustuloksiin. Tämä uudelleenkelpuutusprosessi vaikuttaa tarkoitukselliselta manipuloinnilta haittavaikutusten salaamiseksi turvallisuusanalyysissä, mistä on osoituksena FDA:lle toimitettujen CRF-tiedotteiden suuri osuus asianomaisten koehenkilöiden joukossa.
Lisänäyttöä tietojen manipuloinnista
Haittavaikutusten kirjaamisessa havaittiin muita poikkeamia. Tietojenkäsittelylomakkeiden manuaalisissa tarkasteluissa havaittiin ristiriitaisuuksia, kuten kirjaamattomia haittavaikutuksia ja uudelleen luokiteltuja tai poistettuja tapahtumia. Tämä sisältää merkittävän alikirjaamisen ja CRF-asiakirjojen mahdollisen muuttamisen, mikä rikkoo yleisiä hoitokäytäntöjä ja mahdollisesti Yhdysvaltain ja kansainvälisiä säännöksiä.
Prosessin 2 virheellinen representaatio ja huolenaiheet
Pfizer/BioNTech käytti COVID-19-rokotetta kehittäessään kahta eri valmistusmenetelmää: ”Prosessi 1” suurimmassa osassa kliinisiä tutkimuksia ja ‘prosessi 2’ kaupallisessa tuotannossa, jota testattiin vain 252 vastaanottajalla. Prosessissa 1 käytettiin PCR-monistusta DNA-mallin tuottamiseen, kun taas prosessissa 2 käytettiin linearisoitua plasmidi-DNA:ta, jota kasvatettiin E. coli -bakteerissa, sekä muita tuotantoa lisääviä muutoksia. FDA:n, EMA:n, PMDA:n ja TGA:n sääntelyasiakirjat vahvistavat nämä erot ja niiden käytön kliinisissä ja kaupallisissa toimituksissa.
Suunnitellun vertailututkimuksen tarkoituksena oli arvioida turvallisuutta ja immunogeenisuutta 16-55-vuotiailla osallistujilla, mutta sitä ei lopulta toteutettu. Haittavaikutusraporttien analyysi osoittaa, että prosessien välillä on merkittäviä eroja, erityisesti prosessin 2 vastaanottajilla esiintyi enemmän haittavaikutuksia, kuten lymfadenopatiaa ja menorragiaa. Useat havainnot viittaavat siihen, että prosessien 1 ja 2 väliset valmistusmuutokset ovat vaikuttaneet rokotteen turvallisuusprofiiliin.
Johdanto
COVID-19-rokotteen kehittämisen ja hätätilaluvan (EUA) myöntämisen ennennäkemätön nopeus johti nopeutettuihin tarkistusprosesseihin. Tiukka post-hoc -analyysi on olennaisen tärkeä sen varmistamiseksi, että kaikki tiedot kerättiin, analysoitiin ja raportoitiin korkeimpien tieteellisten standardien mukaisesti. Tämä on ratkaisevan tärkeää tehokkaan markkinoilletuonnin jälkivalvonnan kannalta, sillä tarkat tutkimustiedot ovat olennaisen tärkeitä sellaisten pitkäaikaisvaikutusten tai harvinaisten haittatapahtumien tunnistamisessa, joita ei ehkä ole havaittu alkuperäisten tutkimusten aikana. Virheelliset perustiedot voivat haitata markkinoille tulon jälkeistä seurantaa ja vaarantaa potilasturvallisuuden. Kliinisen tutkimuksen eettiset normit edellyttävät tietojen raportoinnin avoimuutta ja tarkkuutta. Nykyiset puutteet ja avoimuuden puute C4591001-tutkimustiedoissa rikkovat eettisiä periaatteita, kuten tietoon perustuvaa suostumusta, ja ovat mahdollisesti vastoin velvollisuutta olla aiheuttamatta haittaa, jos riski/hyöty on esitetty väärin.
FDA suoritti tarkastelunsa 20. marraskuuta 2020 ja 11. joulukuuta 2020 välisenä aikana, ja tuloksena oli yllättävän lyhyt 57-sivuinen muistio [1]. Tarkastelun vakavuus on asetettu kyseenalaiseksi [2]. Avoimuuslupauksista huolimatta näyttöä useista analyytikkojen suorittamista tarkistuksista, kuten Peter Marksin väittämistä sähköpostiviesteistä, joita olisi vaihdettu niiden data-analyytikkojen välillä, jotka olisivat arvioineet kriittisesti tietoja, ei ole saatavilla. Koska olemme jo yli vuoden ajan etsineet vastauksia olennaisiin tietojen eheyttä koskeviin kysymyksiin, näillä kirjoittajilla on hyvät mahdollisuudet tietää, kuinka huolellisesti Marksin osasto käsittelee näitä huolenaiheita. Muut sääntelyelimet, kuten Ranskan Agence Nationale de Sécurité des Médicaments et des produits de Santé (ANSM) ja Australian Therapeutic Goods Administration, eivät ole toistaiseksi pystyneet vastaamaan asianmukaisesti esitettyihin huolenaiheisiin.
Kun otetaan huomioon välittömät todistajanlausunnot, joissa kuvataan hyvien kliinisten tutkimuskäytäntöjen vakavia rikkomisia, ja tosiseikat, jotka osoittavat, kuinka laajoja väärinkäytöksiä tässä tutkimuksessa esiintyi, on kiireellisesti suoritettava osallistuvien tutkimuspaikkojen ja tiedonhallinnasta vastaavien kliinisten tutkimusorganisaatioiden (CRO) kattava ja avoin tarkastus. Kirjoittajat toivovat, että yleisö hyödyntää tätä toistettavissa olevaa näyttöä, jotta asia saataisiin poliittisten päättäjien tietoisuuteen, ja oikeusviranomaisten on pysyttävä toimettomina.
Tässä kertomuksessa esitetyt tosiseikat perustuvat julkisiin asiakirjoihin, jotka on saatu tiedonvälityksen vapautta koskevien pyyntöjen (FOI) ja oikeudenkäyntien kautta. Merkittävä osa näistä asiakirjoista on peräisin Texasin tuomioistuimen määräyksestä [3] Public Health & Medical Professionals for Transparency (PHMPT) [4] -järjestön aloittamassa menettelyssä, joka mahdollisti sponsoreiden FDA:lle toimittamien tietojen julkaisemisen biologista lupahakemusta (BLA) varten.
Kirjoittajat
Tämä raportti on seuraavien kirjoittajien työn synteesi ja yhteistyön tulos:
Josh Guetzkow, PhD, Vanhempi luennoitsija Jerusalemin heprealaisessa yliopistossa. (Twitter, Substack).
Jeyanthi Kunadhasan MD (UKM), MMed (AnaesUM), FANZCA MMED (Monash) (Twitter, Substack)
Brook Jackson, Kliinisen tutkimusorganisaation (CRO) Ventavian kliinisen tutkimuksen johtaja tutkimuksen aikana, ilmiantaja. (Twitter, Website)
Christine Cotton, Biostatistikko, entinen CRO:n toimitusjohtaja 22 vuoden ajan. (Twitter, Website)
Arkmedic (nimetön), ilmiantaja, PhD & LT (Substack)
Augusto Roux, PhD, lakimies, Buenos Airesin yliopisto, kokeeseen osallistuja, ilmiantaja (Twitter)
Huolestunut amyloidoosi (anonyymi), kliininen tutkija (Twitter, Substack)
OpenVAET (anonyymi), Rikostekninen analyytikko ja data-analyytikko (Twitter, Substack)
Ennakoimamme elementit voidaan tarkistaa riippumattomasti ja käyttää vapaasti. Jos koetta varten tarvitaan allekirjoitettua valaehtoista todistusta, ota meihin yhteyttä kommenttien kautta.
Menetelmä
Viittaukset kaikkiin käytettyihin asiakirjoihin sekä [5] koodi, jota tarvitaan kaikkien edistyneiden lukujen ja kaavioiden jäljentämiseen, ovat mukana.
Latasimme [6], poimimme ja luokittelimme [7] PHMPT:n [8] kautta tällä hetkellä saatavilla olevat tiedot. Jos lähteitä tarvitaan, ne ilmoitetaan alaviitteissä.
Tutkimuksen yleiskatsaus ja demografiset tiedot
Kliinisellä tutkimuksella oli kaksi päätavoitetta:
tuotteen turvallisuus, joka esitetään positiivisen riski-hyötysuhdeprofiilin avulla (toisin sanoen ”säästää enemmän ihmisiä kuin se voi vahingoittaa”)
tuotteen teho, jolla estää oireinen COVID-19-tauti, joka on vahvistettu PCR-testillä FDA:n vaatimusten mukaisesti.
Nämä vaiheet oli suoritettava, jotta saatiin hätäkäyttölupa (EUA). Pfizer ja BioNTech olivat ensimmäiset kilpailevista lääkealan konserneista, jotka ilmoittivat — lehdistötiedotteella 9. marraskuuta 2020 — että nämä tavoitteet oli saavutettu.
EUA:ta sovellettiin 20. marraskuuta 20209 sen jälkeen, kun tiedot oli analysoitu, ja tiedonkeruun päättymispäivä (cut-off) oli 14. marraskuuta 2020. Tämän jälkeen tavoitteena oli saada BLA — tavoite saavutettiin 23. elokuuta 2021 [10] — sen jälkeen, kun hakemus oli jätetty, jonka tietojen rajauspäivä oli 13. maaliskuuta 2021.
Kliinisten lääketutkimusten tietokantaan on rekisteröity yhteensä 48 091 yksilöllistä koehenkilötunnusta, jotka 153 tutkimuspaikkaa seuloi 29. huhtikuuta 2020 ja 12. tammikuuta 2021 välisenä aikana [11].
Tutkimuksessa kullekin tutkimuspaikalle annettiin nelinumeroinen tunniste, joka alkoi numerosta 1001. Yksi tutkimuskohteista, Dr. Mayor Cirujanon sotilaskeskussairaala Argentiinassa, jaettiin kahteen tunnisteeseen, 1231 ja 4444 (virtuaalinen viite [12]). Tutkimuspaikkojen nimet ja osoitteet sekä kunkin tutkimuspaikan päätutkijan henkilöllisyys esitetään yhdessä FDA:lle toimitetussa asiakirjassa [13].
Koehenkilöt rekrytoitiin 10 viikon aikana vaiheen 1 osalta ja 22 viikon aikana vaiheen 2-3 osalta. Seuraavassa kaaviossa esitetään viikoittainen rekrytointi [14].
Koehenkilöiden maantieteelliset koordinaatit yhdistettiin manuaalisesti JSON-tiedostoon [15], joka sisälsi leveys- ja pituusasteet. Kullekin maalle laadittiin yhteenveto suoritetuista rekrytoinneista [16] sekä alla oleva kartta koehenkilöittäin [17].
Tutkimus, joka esiteltiin seuraavasti: ”Plasebokontrolloitu, sokkoutettu tarkkailija, joka osoitti turvallisuuden ja tehon”
Kliininen tutkimus esiteltiin yleisölle ”vaiheen 1/2/3 lumelääkekontrolloituna, satunnaistettuna, sokkoutettuna, annosmittaustutkimuksena SARS-CoV-2 RNA-rokotekandidaattien turvallisuuden, siedettävyyden, immunogeenisuuden ja mahdollisen tehon arvioimiseksi COVID-19-tautia vastaan terveillä henkilöillä”. [18]
Vaiheiden 1 ja 2-3 koehenkilöille tehtiin erilaisia mittauksia (spesifiset neutraloivat vasta-aineet (N-Binding), PCR-testit (PCR), epi:n S1-alayksikölle ja reseptoria sitovalle domeenille (RBD) spesifinen immunoglobuliini G jne.) sen selvittämiseksi, olivatko he saaneet COVIDin ja miten he reagoivat tuotteeseen.
Tehoa mitattiin tuotteen kyvyllä ehkäistä vahvistettuja COVID-19-tapauksia, ja se perustui kahteen ensisijaiseen päätetapahtumaan. Ensisijainen tehokkuustavoite oli estää oireiset laboratoriossa vahvistetut COVID-19-tapaukset vähintään 7 päivää toisen annoksen jälkeen osallistujilla, joilla ei ollut serologista tai virologista näyttöä aiemmasta SARS-CoV-2-infektiosta, ennen rokotusohjelmaa ja sen aikana. Toissijaisena tavoitteena oli arvioida rokotteen tehoa vakavia COVID-19-tapauksia vastaan.
Turvallisuustavoitteisiin kuului paikallisten reaktioiden, systeemisten tapahtumien ja kuumetta alentavien lääkkeiden/kipulääkkeiden käytön arviointi päivästä 1 päivään 7 kunkin annoksen jälkeen osajoukolla osallistujista. Lisäksi tutkimuksessa pyrittiin seuraamaan ei-toivottuja, ei-vakavia haittatapahtumia ensimmäisestä annoksesta kuukauden kuluttua toisesta annoksesta kaikilla osallistujilla, vakavia haittatapahtumia ensimmäisestä annoksesta kuuden kuukauden kuluttua toisesta annoksesta kaikilla osallistujilla sekä kuolemantapauksia ja niihin liittyviä vakavia haittatapahtumia ensimmäisestä annoksesta tutkimuksen loppuun kaikilla osallistujilla. [19]
Vaiheen 1 epäjohdonmukaiset päivämäärät
Kahden vaiheen 1 tuloksista raportoivan tutkimuksen [20, 21] mukaan vaihe 1 aloitettiin 4. toukokuuta 2020, ja rekrytointi (seulonta) jatkuisi 22. kesäkuuta 2020 asti.
Nämä päivämäärät ovat virheellisiä, ja seulonta tapahtui 29. huhtikuuta 2020 ja 29. kesäkuuta 2020 välisenä aikana [22].
Vaihe 1 – Testatut tuotteet ja demografiset näkökohdat
Vaiheen 1 seulontaan valittiin 332 koehenkilöä. 83 ei täyttänyt seulontaehtoja, ja 54:ää ei määritetty hoitoryhmään. Vaiheessa 1 testattiin annostusta ja ensisijaisia turvallisuuskysymyksiä 15 koehenkilön kohorteissa (12:lle annettiin hoitoa ja 3:lle plaseboa). Alun perin testattaviksi suunnitellut hoidot olivat:
BNT162a1 (RNA-LNP-rokote, joka käyttää uRNA:ta ja koodaa RBD:tä): 3 μg, 10 μg, 30 μg
BNT162b1 (BNT162 RNA-LNP-rokote, jossa käytetään modRNA:ta ja joka koodaa RBD:tä): 10 μg, 30 μg, 100 μg
BNT162b2 (BNT162 RNA-LNP-rokote, jossa käytetään modRNA:ta ja joka koodaa P2 S:ää): 10 μg, 30 μg, 100 μg
BNT162c2 (BNT162 RNA-LNP-rokote, jossa käytetään saRNA:ta ja joka koodaa RBD:tä): 3 μg, 10 μg, 30 μg.
Muiden koehenkilöiden oli määrä saada toinen annos 22 päivää annoksen 1 jälkeen – itse asiassa 19-23 päivän [23] kuluttua annoksesta 1. Tämä aikaikkuna olisi EUA:n aikaan ja ilman asianmukaista muutosta tutkimussuunnitelmaan – joka vain mainittiin tilastollisessa analyysisuunnitelmassa – muuttunut 19-42 päiväksi [24].
Vaihe 1 toteutettiin neljässä kohteessa. Ainoastaan Yhdysvalloissa sijaitsevat kohteet 1001, 1002, 1003 ja 1007 olivat mukana [25] — ne on esitetty alla olevassa kartassa [26].
Testattujen tuotteiden määrä oli pienempi kuin alun perin oli suunniteltu:
24 koehenkilöä sai BNT162b1:tä (10 mcg)
12 koehenkilöä sai BNT162b1:tä (20 mcg)
24 koehenkilöä sai BNT162b1:tä (30 mcg)
24 koehenkilöä sai BNT162b2:ta (10 mcg)
24 koehenkilöä sai BNT162b2:ta (20 mcg)
24 koehenkilöä sai BNT162b2:ta (30 mcg)
12 koehenkilöä sai BNT162b1-annoksen (100 mcg), jota seurasi BNT162b1-annos (10 mcg)
39 koehenkilöä sai lumelääkettä
BNT162a1:tä ja BNT162c2:ta ei testattu. Vaiheeseen 1 suunniteltujen koehenkilöiden määrä muuttui toistuvasti [27], kun vaihe 1 oli käynnissä:
840:stä, 17. huhtikuuta 2020, 420:een, 11. kesäkuuta 2020
420:sta 630:een 1. heinäkuuta 2020
630:sta 195:een 24. heinäkuuta 2020.
Vaiheen 1 koehenkilöiden kulku
Tietoon perustuvassa suostumuslomakkeessa [28] annetaan yksityiskohtaiset tiedot koehenkilöiden matkasta vaiheen 1 tutkimuksessa, johon sisältyi yhteensä 10 suunniteltua käyntiä.
Ensimmäisellä tutkimuskäynnillä koehenkilölle tehtiin laaja testisarja, johon kuului myös vasta-ainetasojen mittaaminen sen varmistamiseksi, ettei hän ollut saanut COVID-19-tartuntaa ennen tutkimukseen osallistumista. Myös koehenkilön sairaushistoria käytiin läpi, ja hänen todettiin olevan sopiva osallistumaan tutkimukseen. Tämän seulontaprosessin jälkeen koehenkilö satunnaistettiin ja hänelle annettiin joko annos lumelääkettä tai aktiivista valmistetta.
Seuraavana päivänä, toisella käynnillä, koehenkilön tila vahvistettiin, ja testejä tehtiin vähemmän. Tämä oli ainoa käynti, jossa tutkittavaa ei testattu vasta-aineiden varalta.
Seuraavat käynnit tapahtuivat säännöllisin väliajoin. Kolmas käynti tapahtui viikon kuluttua ensimmäisestä annoksesta, ja neljäs käynti tapahtui toisen annoksen antamisen yhteydessä, joka annettiin 19-23 päivää ensimmäisen annoksen jälkeen. Viides ja kuudes käynti tehtiin viikon ja kahden viikon kuluttua toisesta annoksesta. Seitsemäs käynti tapahtui kuukauden kuluttua toisesta annoksesta, ja kahdeksas käynti oli suunniteltu tehtäväksi kuusi kuukautta myöhemmin. Yhdeksäs käynti oli suunniteltu tehtäväksi vuoden kuluttua toisesta annoksesta, ja kymmenes ja viimeinen käynti oli tarkoitus tehdä kahden vuoden kuluttua toisesta annoksesta.
BNT162b2 100 μg:n koehenkilöt määrättiin erityiseen ”annosteluohjelmaan”, jossa toinen 10 μg:n annos annettiin 85-105 päivää ensimmäisen annoksen jälkeen. Julkisesti saatavilla olevissa pöytäkirjaversioissa ei ole missään dokumentoitu, miksi 100 μg:n koehenkilöitä kohdeltiin näin erityiskohteluun, vaikka tämän olisi pitänyt olla selvää.
Käynnit, joiden vasta-ainemittaukset on säilytetty 13. maaliskuuta 2021 päättymispäivänä vaiheen 1 koehenkilöiden osalta, ovat käynnit 1-8 [29].
Poikkeavuudet vaiheen 1 vasta-ainemittauksissa
Osa koehenkilöiden vaiheen 1 aikana tekemistä vasta-ainemittauksista esitetään Edward Walshin ym. aiemmin mainitussa tutkimuksessa ”Safety and Immunogenicity of Two RNA-Based Covid-19 Vaccine Candidates” [30].
ADVA:n tiedostossa, joka sisältää näiden testien tulokset, on 115 eroavaisuutta [31] samasta koehenkilöstä, saman käynnin aikana, samana päivänä ja saman testin aikana tehtyjen mittausten välillä. Ristiriitatapauksissa pidettiin useimmiten voimassa korkeampi arvo. Vaikka analyysitietojen tarkastajan oppaassa (ADRG [32]) mainitaan tämä poikkeama, siinä ei esitetä syytä siihen. Asiakirjassa, jossa luetellaan analyysimenetelmät [33], on kaksi mNeongreen-viruksen neutralisaatiomääritystä, jotka eroavat toisistaan siten, että toisessa on määritysraja (alaraja ja yläraja pienimmälle ja suurimmalle mitattavissa olevalle titterille). Asiakirjat VR-MQR-10214 [34] ja VR-MVR-10083 [35] ovat ”Qualification” ja ”Method Validation of the SARS-CoV-2 mNeonGreen Virus Microneutralization Assay”, ja niiden toimittamispäivät ovat 18. elokuuta 2020 ja 9. helmikuuta 2021. Vaikka näyttääkin siltä, että uusintatestien jälkeiset erot johtuvat ”validoiduista” neutralisaatiomäärityksen parametreista, kuulemamme asiantuntijat eivät pystyneet selittämään, miten samasta koehenkilöstä samana päivänä otetut näytteet voisivat tuottaa näin merkittäviä eroja mittauksissa – mikä herättää mahdollisuuden, että testit, joiden tulokset olivat ”pettymys”, oli ”uusittu” korvaamalla viallisista koehenkilöistä otetut näytteet näytteillä, joiden vasta-ainepitoisuudet olivat tunnetusti korkeat. Jos menetelmän muutos olisi pätevä, se herättäisi joka tapauksessa kysymyksiä aiempien mittausten pätevyydestä.
Esimerkkinä alla oleva kaavio havainnollistaa ”SARS-CoV-2-seerumin neutraloiva titteri 50 (titteri) — virusneutralointimääritys” -mittausten eroja, kun verrataan ensimmäisten tehtyjen mittausten keskiarvoa ja jatkuvien mittausten keskiarvoa.
Vaiheen 2-3 koehenkilöiden kulku
Tietoon perustuvassa suostumuslomakkeessa annetaan yksityiskohtaiset tiedot koehenkilöiden matkasta vaiheen 2-3 tutkimuksessa [36], johon kuului yhteensä kuusi suunniteltua käyntiä.
Ensimmäisellä käyntikerralla koehenkilölle tehtiin pitkä luettelo testejä, vaikkakin vähemmän kuin vaiheen 1 koehenkilöiltä vaadittiin. Näihin kuului muun muassa koehenkilön vasta-ainetasojen mittaaminen sen varmistamiseksi, ettei hän ollut saanut COVID-19-tartuntaa ennen tutkimukseen osallistumista. Tutkittavan sairaushistoria käytiin läpi, ja hänen todettiin olevan sopiva osallistumaan tutkimukseen. Tämän jälkeen koehenkilö satunnaistettiin ja hänelle annettiin joko lumelääkeannos tai vaiheen 1 jälkeen ylläpidetty aktiivinen tuote, BNT162b2 30 mcg.
Toisella käynnillä annettiin toinen annos. Tämä oli ainoa käynti, jolloin ei tehty vasta-ainemittauksia, jotka olisivat voineet antaa vahvistuksen infektioista, joita on voinut esiintyä ensimmäisen annoksen jälkeisinä päivinä oireettomina tai jotka ovat välttyneet PCR-tunnistukselta esimerkiksi siksi, että koehenkilö ei ole ilmoittanut oireistaan tai että häntä ei ole testattu oireiden ilmoittamisen aikana.
Kolmas käynti tapahtui kuukauden kuluttua toisesta annoksesta, ja neljäs käynti oli suunniteltu pidettäväksi 6 kuukauden kuluttua toisesta annoksesta. Viides käynti tapahtui vuoden kuluttua toisesta annoksesta, ja kuudes ja viimeinen käynti oli suunniteltu tehtäväksi kahden vuoden kuluttua toisesta annoksesta.
Ilmoitettujen satunnaistamislukujen ristiriitaisuudet
Toimeksiantajat tuottivat kaksi ”tutkijatiedostoa”, joissa raportoitiin tutkimuskohteiden tutkijat ja kussakin kohteessa seulottujen ja satunnaistettujen koehenkilöiden kokonaismäärä. Ensimmäinen asiakirja laadittiin 26. marraskuuta 2020 [37] ja toinen 29. maaliskuuta 2021 [38]. Ensimmäisessä asiakirjassa mainitaan kyselypäiväksi 19. marraskuuta 2020 ja toisessa 19. maaliskuuta 2021. Vaikka seulottujen koehenkilöiden kokonaismäärässä toimipaikkaa kohti ei ole suurempaa muutosta kuin toimipaikkaa vaihtaneiden koehenkilöiden määrässä, satunnaistettujen koehenkilöiden kokonaismäärä laskee merkittävästi [39] ilman selitystä. Koehenkilöitä ei voida ”poistaa satunnaistamisesta” kliinisessä tutkimuksessa.
Esimerkiksi alla olevassa kuvassa Argentiinassa sijaitseva koehenkilö 1231/4444 muuttui 5776 satunnaistetusta koehenkilöstä 26. marraskuuta 2020 5615:een (-161) 29. maaliskuuta 2021.
Kaikkiaan 1203 koehenkilöä putosi pois tutkimuksesta, mikä vastaa 108:aa 153:sta tutkimuskohteesta. Tämä on merkittävä poikkeama, jota ei voida selittää koehenkilöiden siirtymisellä toimipaikkojen välillä tai alle 16-vuotiaiden koehenkilöiden rekrytoinnilla, jotka olisi sisällytetty yhteen raporttiin eikä toiseen. Anomalian havainnollistamiseksi voidaan todeta, että tutkimuspaikka 1018 menetti 30 koehenkilöä, kun taas yksikään koehenkilö ei siirtynyt sisään tai ulos ja että yhtään alle 16-vuotiasta nuorta ei ollut rekrytoitu 19. marraskuuta 2020, jolloin ensimmäinen tiedosto kysyttiin – eikä missään vaiheessa sen jälkeen. Alla oleva ote havainnollistaa kaikkia koepaikkoja, jotka menivät satunnaistamisluvuissa taaksepäin [40].
Vaihe 2-3 – Satunnaistamisluvut virallisesti julkaistu
ADRG [41] dokumentoi 6 koehenkilöä, jotka rekisteröityivät useammalle kuin yhdelle koepaikalle, mikä synnyttää 12 koehenkilötunnusta ja joihin sovelletaan tiettyjä poissulkemisia (10561101, 11331382, 11101123, 11331405, 11491117, 12691090, 12691070, 11351357, 11341006, 10891112, 11231105, 10711213).
BLA-hakemusmuistiossa [42] ja vastaavassa Thomas et al. NEJM-tutkimuksessa ”Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine through 6 Months ” [43] ilmoitetaan molemmissa yhteensä 44 165 satunnaistettua koehenkilöä 13. maaliskuuta 2021 mennessä. Tämä populaatio on toistettu [44] ja sen vastaavuus ilmoitettujen demografisten ominaisuuksien kanssa on varmistettu [45].
Sokkokokeeseen liittyvät poikkeamat
Potilaiden, joiden käyttäytyminen poikkesi tutkimussuunnitelmassa esitetystä tai joiden kohdalla tutkimusryhmä teki virheen, tutkimussuunnitelmapoikkeamat kirjattiin ylös, minkä jälkeen erillinen ryhmä tarkasteli niitä uudelleen määrittääkseen, oliko poikkeama niin vakava, että se oikeuttaisi potilaan sulkemisen pois tietystä ”populaatiosta” (ryhmä, joka oli tarkoitettu jonkin tutkimustavoitteen (esim. tehon) analysointiin).
Sääntelyviranomaisten hyväksymä tutkimussuunnitelma [46] poikkesi merkittävästi tavanomaisesta kliinisen tutkimuksen käytännöstä: paikan päällä työskentelevää henkilökuntaa, joka oli vastuussa tutkimussuunnitelmasta poikkeamisen tutkimisesta ja osallistujien mahdollisen poissulkemisen määrittämisestä, ei tarkastettu sokkona.
Satunnaistetun vaiheen 3 populaation [47] tuottamien poikkeamien analyysi tuo esiin merkittäviä epäsuhtaisuuksia hoitoryhmissä. Seuraavassa taulukossa on yhteenveto kaikista poikkeamista, jotka havaittiin yli 100 tutkimushenkilöllä ja jotka osoittavat tilastollisesti merkittävää epätasapainoa verrattuna satunnaistetun populaation muuhun osaan (22 085 BNT162b2 & 22 080 Placebo), käyttäen khiin neliö -testiä.
Eräät hyvin merkittävät poikkeamat — kuten ”Minkä tahansa muun kuin tutkimukseen kuuluvan koronavirusrokotteen saaminen milloin tahansa ennen tutkimusta tai sen aikana” — osoittavat, että monet koehenkilöt olivat tietoisia siitä, että he olivat saaneet lumelääkettä — ja olivat samanaikaisesti osallistuneet kilpailijan tutkimukseen (todennäköisesti Moderna) tai saaneet toisen tuotteen hätätilanteessa myönnetyn käyttöluvan jälkeen. Toisaalta tietyt epäsuhtaisuudet osoittavat, että tiettyjen toimipaikkojen tutkimusryhmät eivät kohdelleet potilaita siten kuin sokkoutetussa havainnointitutkimuksessa odotetaan, jossa kliinisen tutkimusryhmän olisi kohdeltava molempia ryhmiä tasapuolisesti:
Akuutin reaktion arviointi protokollassa määritellyn ajanjakson aikana tutkimustoimenpiteen antamisen jälkeen, jota ei suoriteta rokotuskäyntien yhteydessä
Hoitopaikan henkilökunta ei ole ottanut nenänäytettä ennen rokotusta
Nenänäytettä ei ole otettu sitä käyntiä varten, jota varten sitä tarvitaan
Toimenpide/testi, jota ei ole tehty protokollan mukaisesti
Raskaustestiä ei ole tehty virtsasta.
Poikkeamien ”Minkä tahansa muun kuin tutkimuksessa käytetyn koronavirusrokotteen saanti” osalta on syytä huomata, että suuri osa niistä tapahtui joulukuun 2020 jälkeen, kun muita rokotteita tuli saataville.
Kymmenen koehenkilöä, joita koskee 20 tai enemmän näistä poikkeamista ja jotka osoittavat merkittävää epätasapainoa Fisherin tarkalla testillä testattaessa, on eristetty ja koottu seuraavaan taulukkoon.
Toisin kuin 32 kohdassa tarkoitetut ”koehenkilöihin liittyvät” poikkeamat, 31 kohdassa mainitut ”henkilökuntaan liittyvät” poikkeamat tutkitaan suurimmaksi osaksi ennen koehenkilöiden virallista sokon purkamista.
COVID-käynnit ja oireiden poikkeavuudet
Kun koehenkilöt kokivat COVID-oireita FDA:n ennalta määrittelemästä luettelosta, heidän oli mentävä tutkimuspaikalle ottamaan näytteitä — tai, jos se ei onnistunut, testattava itsensä kotona toimitetuilla testeillä ja lähetettävä näyte tutkimuspaikalle neljän päivän [48] kuluessa ennen oireiden alkamista tai sen jälkeen. ”Viralliset” COVID-oireet olivat seuraavat 13 oiretta [49]:
Vilunväristykset
Ripuli
Kuume
Haju- tai makuaistin katoaminen
Uusi tai pahempi yskä
Uusi tai pahempi lihaskipu
Uusi tai pahempi hengenahdistus
Kurkkukipu
Oksentaminen
Näiden oireiden lisäksi oli myös ”epävirallisia” oireita:
Väsymys
Päänsärky
Nenän tukkoisuus
Vuotava nenä
”Covid-19 Signs and Symptoms” .XPT-tiedostossa (ADSYMPT [50]) luetellaan kuitenkin yhteensä 15 oiretta — ne, joita ei ole merkitty protokollaan, on merkitty (*):
Vilunväristykset
Ripuli
Väsymys
Kuume
Päänsärky
Pahoinvointi (*)
Haju- tai makuaistin katoaminen
Uusi tai pahempi yskä
Uusi tai pahempi lihaskipu
Uusi tai pahempi nenän tukkoisuus
Uusi tai pahempi hengenahdistus
Uusi tai pahempi kurkkukipu
Uusi tai pahempi hengityksen vinkuna (*)
Vuotava nenä
Oksentaminen
Tutkimuspaikoilla oli myös mahdollisuus ilmoittaa ”muita” oireita, joita ei kuitenkaan kirjattu tietokantaan. Esimerkiksi koehenkilö 10161289 [51], 17-vuotias mies, BMI 22,4, joka sai BNT162b2 30 mcg -annoksen 1 18. syyskuuta 2020 ja annoksen 2 7. lokakuuta 2020. Hänellä oli COVID 7. ja 11. lokakuuta, arviointi 9. lokakuuta ja oireet: kuume, yskä, lihaskipu, kurkkukipu, nuha. Tapausselostuslomakkeiden sivulla 270 ”rintakipu” on kuitenkin mainittu ”muuna” oireena. Sitä ei ole raportoitu haittavaikutuslokissa, ja se on näkyvissä vain siksi, että nuorella miehellä oli myöhemmin 31. lokakuuta 2020 vakava haittavaikutus : moottoriajoneuvo-onnettomuus, jossa hän kärsi kasvojen murtumia ja aivotärähdyksen, mikä johti hänen CRF:nsä julkaisemiseen. Tämä on vastoin yleisiä terveyssääntöjä – myös EMAn hyvää kliinistä käytäntöä koskevia ohjeita [52].
Toinen esimerkki siitä, että hoitopaikat ovat rikkoneet GCP:tä, on koehenkilö 12315632 [53], toinen BNT162b2 30 mcg -vastaanottaja. Hänen COVID-sairausmerkintänsä kattaa ajanjakson 17. lokakuuta 2020 – 7. joulukuuta 2020, ja siihen sisältyy kaksi sairaalahoitoa. Vaikka hoitopaikka kuvailee hänen tilansa aluksi keuhkokuumeeksi, koehenkilö kärsi SAE:stä (sydänpussitulehdus), joka raportoitiin vasta sponsorin pyynnöstä. hoitopaikka raportoi aluksi ”keuhkokuumeen” haittatapahtumana, mutta poisti sen myöhemmin ja siirsi sen COVID-sairauskäyntiin. Jos Pfizer ei olisi vaatinut sydänpussitulehduksen merkitsemistä sydänpussitulehduksen haittavaikutusrekisteriin, se olisi jäänyt raportoimatta tutkimustiedoissa ja olisi ollut vain Pfizerin faksilla lähetetyssä turvallisuustietokannassa.
Sokkouttamisen ja hyvien kliinisten käytäntöjen kunnioittamatta jättäminen, Brook Jacksonin lausunto
Ilmiantaja Brook Jackson, joka toimi alueellisena johtajana Ventavia Research Groupissa (Ventavia), joka oli tutkimukseen osallistujien rekisteröinnistä vastaava toimipaikan hallinnointiorganisaatio, toimitti British Medical Journalille (BMJ) yrityksen sisäisiä asiakirjoja, valokuvia, äänitallenteita, sähköpostiviestejä ja muuta todistusaineistoa, joka herätti huomattavia huolenaiheita tietojen eheydestä ja tutkimukseen osallistujien turvallisuudesta [54].
Hän raportoi muun muassa seuraavista eri tutkimuspaikoissa dokumentoiduista järjestelmällisistä ongelmista:
Asianmukaisen tietoon perustuvan suostumuksen puuttuminen: Sisäisessä laadunvarmistustarkastuksessa, jossa tarkasteltiin satoja yksittäisiä osallistujatietoja, havaittiin, että jokaiselta osallistujalta ei saatu tietoista suostumusta ennen tutkimusvalmisteen pistämistä. Lisäksi todettiin, että monissa näistä lomakkeista oli selviä allekirjoitusvirheitä, jotka viittasivat potilaan allekirjoitusten väärentämiseen.
Kelpaamattomien kliinisen tutkimuksen osallistujien rekisteröinti: Kliiniseen tutkimukseen otettiin mukaan osallistujia, jotka eivät täyttäneet Pfizerin asettamia sisäänottokriteerejä tai jotka täyttivät poissulkukriteerit. Tämä koski raskaana olevia naisia, Ventavian työntekijöitä, perheenjäseniä ja ystäviä.
Tietojen väärentäminen ja tietojen väärentäminen: Asiakirjat osoittivat, että datapisteitä oli väärennetty ja väärennetty. Esimerkkejä ovat:
Potilastietojen muuttaminen ja asiakirjojen takautuvuus.
Lääkärin allekirjoitusten väärentäminen.
Puuttuvien tai epäjohdonmukaisten tietojen väärentäminen, mukaan lukien sairaushistoria, samanaikainen lääkkeiden käyttö, fyysiset tutkimukset, elintoiminnot, verenkeräys- ja näytteiden käsittelyajat, virtsan raskaustesti ja muut olennaiset tutkimustiedot.
Lainsäädännön noudattamatta jättäminen/muut protokollarikkomukset
”Sokkoa” ei ole pidetty yllä vaaditulla tavalla.
Protokollaan vaaditut tutkimuskäynnit, mukaan lukien mahdolliset COVID-19-sairauskäynnit, joita ei ole tehty tai joita ei ole suoritettu määrätyn aikataulun ulkopuolella.
Osallistujien sähköisen päiväkirjasovelluksen käytön seuraamatta jättäminen, jota käytettiin paikallisten reaktioiden, systeemisten tapahtumien ja injektion jälkeisen kuumetta alentavan lääkityksen käytön kirjaamiseen.
Haittatapahtumien ja vakavien haittatapahtumien selvittämisen, keräämisen ja raportoinnin laiminlyönti, mukaan lukien tapahtuman tai sen seurausten seuranta.
Tutkimusvalmisteen annostelu- ja antovirheet.
Tutkimusvalmisteen lämpötilan asianmukaisen valvonnan laiminlyönti.
Osallistujien laboratorionäytteiden virheellinen merkitseminen ja sekoittaminen.
Epäpätevän ja kouluttamattoman tutkimushenkilöstön käyttö, mukaan lukien sokkona toimivat rokottajat ja laboratoriohenkilöstö.
Päätutkijan valvontahäiriöt.
Riittämätön hätäapuprotokolla akuutin allergisen reaktion sattuessa.
Rokotuksen jälkeisten akuuttien reaktioiden arvioinnin laiminlyönti.
Vakavien haittatapahtumien, protokollan ja säännösten rikkomisen ilmoittamatta jättäminen Pfizerille tai ulkoiselle institutionaaliselle tarkastuslautakunnalle.
Raskauden, imetyksen ja työperäisen altistumisen aikana tapahtuvan tutkimusvalmistealtistuksen ilmoittamatta jättäminen ja seurannan laiminlyönti.
Muut eettiset rikkomukset, mukaan lukien HIPAA:n rikkominen ja hyväksymättömän korvauksen maksaminen, jotta vältettäisiin osallistujien valitukset FDA:lle tai tiedotusvälineille.
Ilmoitettuaan Ventavialle toistuvasti näistä huolenaiheista ilman, että mitään toimenpiteitä olisi toteutettu tutkimukseen osallistuneiden oikeuksien, turvallisuuden, hyvinvoinnin ja yksityisten terveystietojen suojelemiseksi, hän teki valituksen Center for Biologics Evaluation and Research (CBER) -virastolle, joka on FDA:n osasto, joka sääntelee ihmisille tarkoitettuja biologisia tuotteita sovellettavien liittovaltion lakien, kuten Public Health Service Actin ja Federal Food, Drug and Cosmetic Actin, mukaisesti. Ventavia irtisanoi hänet muutamassa tunnissa CBER:n ilmoittamisen jälkeen.
FDA:n biotutkimuksen valvontaohjelma (BIMO) on ohjelma, johon kuuluu tarkastuksia paikan päällä, tietojen tarkastuksia ja sääntelyn etäarviointeja. Sen tarkoituksena on valvoa kaikkia FDA:n sääntelemien tutkimusten toteuttamiseen ja raportointiin liittyviä näkökohtia. BIMO-ohjelman tavoitteena on varmistaa FDA:lle uusien tuotteiden hyväksyntää ja markkinointihakemuksia varten toimitettujen tietojen laatu ja eheys ja samalla suojella tutkimukseen osallistuvien tutkittavien oikeuksia ja hyvinvointia [55]. Vaikka FDA:n BIMO-ohjelma sai valituksen, jossa ilmoitettiin merkittävistä rikkomuksista, se ei tarkastanut näitä kriittisiä paikkoja. Sen sijaan tarkastuksia tehtiin kuudessa muussa tutkimukseen osallistuneessa kliinisen tutkimuksen toimipaikassa. FDA:n tarkastusmuistion mukaan näissä tarkastuksissa ei havaittu ongelmia, jotka olisivat vaikuttaneet alkuperäisen EUA:n hyväksynnän tueksi toimitettuihin tietoihin.
Augusto Roux’n todistus: Haitallisten vaikutusten kirjaamatta jättäminen ja uudelleenarviointi
Yksi Argentiinassa tehtyyn kliiniseen tutkimukseen osallistuneista, asianajaja Augusto Roux, on myös tämän raportin kirjoittajien joukossa. Hän raportoi, että kokeen aikana tapahtui useita vakavia rikkomuksia yleisiä toimintaperiaatteita ja potilasturvallisuutta vastaan. Tohtori David Healy, kanadalainen yliopistonlehtori ja lääkäri, kertoi tapahtumista yksityiskohtaisesti verkkosivustollaan [56] — sitten vertaisarvioidussa artikkelissa ”The coverage of medical injuries in company trial informed consent forms ” [57].
Augusto Roux ilmoittautui vapaaehtoisesti tutkimukseen, jossa hänelle annettiin koehenkilönumero 12312982, ja hän sai ensimmäisen annoksensa 21. elokuuta 2020. Hän sai toisen annoksensa 9. syyskuuta 2020 klo 18.00. Toisen annoksen jälkeen hän tunsi itsensä hyvin huonovointiseksi puolitoista tuntia myöhemmin, lyyhistyi kotiin päästyään ja sai kaksi päivää myöhemmin Alemánin sairaalassa diagnoosin sydänpussitulehduksesta, jonka useat lääketieteen asiantuntijat arvioivat syy-yhteydeksi hänen saamaansa rokotteeseen: tohtori David Healyn lisäksi tohtori Gemma Torrell, barcelonalainen perhelääketieteen erikoislääkäri ja professori Joan-Ramon Laporte, yksi Espanjan johtavista lääketurvatoiminnan asiantuntijoista.
Augusto Roux otti yhteyttä FDA:han, EMA:han ja muihin sääntelyviranomaisiin, jotka eivät käsitelleet hänen tapaustaan sen edellyttämällä vakavuudella. Hän käynnisti Argentiinassa rikostutkinnan Fernando Polackia ja muita vastaan julkisten asiakirjojen väärentämisestä ja henkilöstä luopumisesta.
Hänen lääkärinsä antama asiantuntijalausunto on tämän kertomuksen liitteenä [58]. Nämä kolme asiantuntijaa ovat muiden vakavien yleisten hoitokäytäntöjen rikkomisten ohella raportoineet seuraavaa:
Hänen haittavaikutuksistaan ei raportoitu oikein – ja osa niistä luokiteltiin myöhemmin uudelleen mahdolliseksi COVID:ksi sponsorin, BioNTechin, pyynnöstä.
Esimerkiksi sydänpussitulehdusta ei yksinkertaisesti raportoitu [59].
Hänen lääkäreilleen toimitetut kliinisten tutkimusten tiedot sisälsivät useilta potilailta kerättyjen näytteiden sekoituksia, ne eivät olleet aikajärjestyksessä ja rikkoivat muita hyviä käytäntöjä.
Lääkäreille toimitetut tiedot vaikuttivat sekavilta ja puutteellisilta.
Asiakirjoista käy ilmi, että tohtori Fernando Polack yritti ilman alan asiantuntemusta luokitella Augusto Roux’n mielisairaaksi ja salata vamman ja rokotteen välisen yhteyden.
Puhelintallenteet viittaavat siihen, että kuolemantapaukset, jotka olisivat tapahtuneet kokeen aikana, olisi salattu.
Lisäindikaattorit koehenkilöiden poistamisesta tietokannasta, 301:sta puuttuvasta koehenkilötunnistetiedosta
Kun koehenkilö rekisteröitiin, ICON-ohjelmisto (”Firecrest”, Oracle-keskustietokannan synkronoima käyttöliittymä) antoi sille yksilöllisen ja ennen kaikkea inkrementaalisen tunnisteen (”koehenkilön id” – skalaari ”SUBJID” .XPT-tiedostoissa):
Neljä ensimmäistä numeroa on tutkimuspaikan tunnus, ja ne ovat aina samat kaikille saman tutkimuspaikan koehenkilöille. (Joissakin tapauksissa koehenkilöt vaihtoivat tutkimuspaikkaa tutkimuksen aikana, joten koko yksilöllinen koehenkilötunnus antoi tietoa sekä ”nykyisestä” että ”alkuperäisestä” tutkimuspaikasta).
Neljä viimeistä numeroa osoitti rekisteröintijärjestyksen, joka alkoi numerosta 1001. Seuraava koehenkilö, joka rekisteröitiin tietylle testauspaikalle, sai numeron 1002, sitten 1003 ja niin edelleen.
Tutkimuksen tunniste on ympyröity sinisellä (aina C4591001), nykyisen tutkimuspaikan tunniste on ympyröity vihreällä (tässä 1016). Alkuperäinen rekisteröintipaikka on ympyröity keltaisella. Kyseisen paikan lisääntyvän tutkimushenkilön tunniste on ympyröity punaisella.
Näiden numeroiden — jotka ovat yleensä täysin peräkkäisiä — analyysi [60] osoittaa, että 301 koehenkilöä puuttuu toimitetuista raporteista. Tämä poikkeama vaikuttaa suhteettomasti Argentiinaan, jossa ainoa koepaikka edustaa 111:tä näistä 301:stä puuttuvasta koehenkilöstä.
17 näistä koehenkilöistä — yksi merkittävimmistä eroista yhdellä koepaikalla ja yhtenä päivänä — rekrytoitiin samana päivänä kuin Augusto Roux, 21. elokuuta 2020.
Jos puuttuvat koehenkilötunnukset johtuisivat satunnaisvirheestä, olettaisimme, että puuttuvat tunnukset jakautuisivat tasaisesti kaikkiin tutkimuskohteisiin. Analyysimme osoittaa kuitenkin, että kuvio ei ole satunnainen, vaan puuttuvia tunnistetietoja on suhteettoman paljon vain muutamissa toimipaikoissa. Lisäksi odotimme havaitsevamme yksittäisiä puuttuvia tunnistenumeroita, mutta sen sijaan havaitsimme usein peräkkäisten puuttuvien tunnistenumeroiden klustereita, joissa puutteet vaihtelevat 2:sta 9:ään tai useampaan peräkkäiseen tunnistenumeroon. Seuraavassa jaottelussa esitetään yhteenveto erikokoisten aukkojen yleisyydestä.
Jos nämä puuttuvat koehenkilötunnisteet johtuisivat jostain tietokonevirheestä (vaikka se olisi kuinka epätodennäköistä, koska olemme hyvin kaukana sellaisista määristä, jotka vaikuttaisivat Oraclen tietokannan välimuistiin), odottaisimme, että niitä esiintyisi kaikkein kuormitetuimpina päivinä. Voimme esittää koepaikalle 1231 rekisteröidyn päivittäisen seulonnan ja koehenkilöt, jotka ovat saattaneet kadota päättämillämme rekrytointipäivillä, havainnollistamaan sitä, että 21. elokuuta 2020 ei ollut tämän koepaikan koehenkilöiden ilmoittautumisen kuormitetuin päivä.
Epäjohdonmukaisuudet välivaiheen tehokkuusanalyyseissä: ilmoituspäivämäärät ja yhteensopimaton koehenkilö
SAP-ohjelmassa [61] tehoanalyysi tehtiin 164 tapauksessa EU:n tehokkuusselvitystä varten ja välivaiheen tehoanalyysit 62 ja 92 tapauksessa, jolloin teho voitaisiin ilmoittaa, jos se saavutetaan.
Yhdessä toimitetuista .PDF-tiedostoista [62] on lueteltu 170 ”virallista” tehokkuustapausta, jotka on esitetty 14. marraskuuta 2020 tapahtuneena pysäytyspäivänä. Tapaukset eivät sisällä niiden keskeistä PCR-päivämäärää (jolloin Pfizer sai tiedon ja vahvistuksen tapauksesta). Näiden tapausten poimiminen [63] mahdollistaa niiden uudelleen analysoinnin [64].
Se osoittaa myös, että Pfizerilla oli 22. lokakuuta 2020 jo tarvittavat 63 tapausta ja 30. lokakuuta 2020 jo 94 tapausta. Siitä huolimatta se odotti 9. marraskuuta 2020 asti ilmoittaakseen väliaikaisen tehokkuusanalyysin tulokset lehdistötiedotteella. On epäselvää, miksi he viivyttelivät tuotteen väitetyn tehon ilmoittamista niin kauan. Ehkä se liittyy siihen, että rokotteen saaminen oli keskeinen kysymys 3. marraskuuta 2020 pidetyissä Yhdysvaltain vaaleissa — ja Pfizerin toimitusjohtajan Albert Bourlan [65] ilmaisemaan haluun olla politisoimatta keskustelua entisestään.
Poikkeamat EUA-tehokkuustapauksissa
Kuten tohtori Jeyanthi Kunadhasan korosti australialaisessa The Spectator -lehdessä julkaistussa artikkelissaan [66] , jossa hän raportoi työstään DailyCloutin ryhmän 3 kanssa, useat merkittävät protokollapoikkeamat ovat vaikuttaneet EUA:n virallisiin tapauksiin sisältyviin koehenkilöihin:
170 osallistujasta viisi (4 BNT ja 1 lumelääke) ei saanut toista annosta protokollan määrittelemän 19-23 päivän ajanjakson kuluessa. Kuten kohdassa 13 mainittiin, 19-42 päivän aikaväli mainittiin vain EUA:n asiakirjoissa [67].
Yksi osallistuja (lumelääke) ei saanut oikeaa annosta valmistetta.
Toinen osallistuja (lumelääke) sai verivalmistetta 60 päivän kuluessa, mikä oli merkittävä poikkeama tutkimussuunnitelmassa.
Yksi osallistuja (44441224, lumelääke [68]) vetäytyi tutkimuksesta ennen EUA:n myöntämispäivää, mutta oli edelleen mukana 170:ssä.
Vasta-ainemittaukset & poikkeavuudet sokkokokeessa
N-vasta-aineita tutkittiin testeillä, jotka reagoivat SARS-CoV-2 -viruksen nukleokapsidiksi (N) kutsuttuun molekyyliin. Anti-N-vasta-aineet ovat erityisen kiinnostavia, koska ainoa tunnettu tapa, jolla tutkittava voi tuottaa niitä, on COVID-19-tartunta. N-vasta-aineet mitattiin tutkimuksen aikana mNeonGreen SARS-CoV-2 ”Nucleoprotein-Binding Antibodies Assay ” [69] -menetelmällä. Kuten nimestä voi päätellä, tämä testi kohdistuu nukleoproteiiniin, joka on nukleokapsidin osa [70]. On syytä huomata, että N-vasta-aineet eivät ole täydellinen havaitsemismenetelmä, sillä havainnointitutkimusten mukaan 5,5 prosenttia tutkittavista oli negatiivisia 100 päivän kuluttua [71].
Lumelääkehenkilöillä oli huomattavasti suurempi todennäköisyys olla osallistumatta tutkimuskäynnille 3 tai jättää vasta-aineet testaamatta tutkimuskäynnillä 3, kuten N-sitoutumisen [72] kautta tehdyt vasta-ainetestaukset osoittivat.
21:llä 1178:sta puuttuvasta käynnin 3 BNT:stä on positiivinen keskitetty PCR (1,78 %), ja 105:llä puuttuvasta 1392:sta lumelääkehenkilöstä on positiivinen PCR (7,5 %) (Käynti 3 suoritettiin 46-138 päivää käynnin 1 jälkeen). COVID:n havaitseminen näyttää olleen tekijä, joka vaikutti siihen, että koehenkilöt eivät olleet käyneet suunnitellulla käynnillä 3. Se ei kuitenkaan riitä selittämään kuittausta — sillä vaikka nämä positiiviset PCR-testit jätettäisiin pois, ero on edelleen merkittävä.
21 191 BNT- ja 21 191 lumelääkehenkilöä aloitti siten, kun PCR- ja N-sitoutumistulokset olivat negatiiviset käynnillä 1, ja he saivat ensimmäisen annoksensa. 19 928 BNT- ja 19 511 lumelääkehenkilöä olivat negatiivisia N-sitovien vasta-aineiden testauksessa käynnillä 3, ja 85 BNT-henkilöä oli positiivisia, kun taas 288 lumelääkehenkilöä oli positiivisia. 53 BNT-tapausta havaittiin keskitetyn PCR:n avulla, ja 201 Placebo-tapausta. BNT-henkilöiden joukossa oli 32 havaitsematonta tapausta (37,6 % 85 tapauksesta), kun taas plaseboryhmässä oli 87 havaitsematonta tapausta (30,2 %) — tämä osoittaa, kuinka virheellinen PCR-tunnistus oli ja kuinka valitettavaa on, ettei vasta-aineita ole mitattu käynnillä 2. Tämä osoittaa, että PCR-tunnistus oli virheellinen ja että on valitettavaa, ettei vasta-aineita ole mitattu käynnillä 2. BNT-ryhmiin kuuluvilla henkilöillä oli 7,4 % suurempi todennäköisyys välttää PCR-tunnistus, mutta heillä oli positiivisia COVID-spesifisiä vasta-aineita 1 kuukauden kuluttua annoksen 2 jälkeisestä käynnistä.
Kommentit-tiedostossa (CO.xpt) on 9113 kommenttia, jotka liittyvät COVID-verinäytteiden ottamiseen toipilaiden käynneillä (tunnistetiedot RDOMAIN=IS ja VISIT=COVID_n), ja 8869 toipilaiden käyntiä toipilaiden käynneillä (SV.xpt) on tiedostossa Subject visits (SV.xpt) (tunnistetiedot VISIT=COVID_n). Kuitenkin vain 91 tulosta liittyy COVID-kävelyihin toipilaana IS.xpt (Immunogeenisuus) -tietokannassa (tunnistettu VISIT=COVID_n1). Protokolla sallii päällekkäiset käynnit; jos yksi käynneistä oli toipilaskäynti, näytteet oli tarkoitus kirjata sinne. Toipilasajan serologiset tulokset jäivät raportoimatta, ja vaikka selitys käynnin 3 puuttumiselle voi johtua siitä, että lumelääkehenkilöt olivat menettäneet kiinnostuksensa tutkimukseen, tämä on protokollavirhe, joka mahdollisti tietojen raportoimatta jättämisen.
Haitalliset vaikutukset COVID-käynnin uudelleenkvalifiointiin olivat laajalle levinneet.
Augusto Roux’n todistajanlausunto, kohdat 43-46, on todiste siitä, että protokollan heikkoutta — joka koostui COVID-oireiden ja tuotteen haittavaikutusten sekoittamisesta keskenään sekä kyvystä luokitella jompikumpi niistä vaikuttamaan teho-, turvallisuus- ja immunogeenisuustuloksiin — käytettiin hyväksi.
Lukuun ottamatta yhtä riviä, joka koskee kahta tutkittavaa (10681017 ja 10681036), kaikissa haittavaikutuksia koskevissa merkinnöissä on 1-N merkintää, jotka liittyvät tutkittavaan yksilöllisen lisäysmerkinnän ”AESPID ‘ [73] avulla, joka on dokumentoitu CSDRG:ssä [74] ’sponsorin tunnisteeksi tietyille haittavaikutuksille” (Sponsor’s ID set for the specific AEs). Sponsoreiden asiakirjassa 5 poikkeusta oli nostettu esiin 13. maaliskuuta 2021, koska toimipaikat olivat syöttäneet tässä asiakirjassa päällekkäisiä tunnuksia.
Analysoimalla tietorakennetta, jotta voidaan havaita aukkoja näissä AESPID-tiedoissa koehenkilökohtaisesti, voidaan havaita haittavaikutukset, jotka poistettiin ja jätettiin pois turvallisuusanalyysistä: vähintään 1209 haittavaikutusta on uudelleen kelpuutettu (R) [75], 767 koehenkilön osalta. Lokin lopussa olevat haittatapahtumat välttävät havaitsemisen.
Rajoitetun määrän koehenkilöitä (yhteensä 1028 koehenkilöä, joista 982 yksittäisissä tiedostoissa) osalta CRF on toimitettu FDA:lle BLA-hakemuksen mukana. Näistä yksilöllisistä CRF-tiedostoista 97 koskee 767 koehenkilöä (12,65 %). Tämä erittäin merkittävä osuus saatavilla olevista CRF:istä, kun taas yli 44K:n populaatiosta on saatavilla vain 2 %, saattaa selittyä sillä, että FDA:lla on kysymyksiä erityisesti osasta näistä tutkittavista — tai yksinkertaisesti sillä, että he noudattavat CRF:ien toimittamiselle asetettuja ehtoja, jotka on eritelty kokousten kirjeenvaihdossa [76].
Koehenkilön kirjausketjun avulla voimme tunnistaa, mitkä haittavaikutukset poistettiin rekisteröinnin jälkeen, kun tapahtumat ”kelpuutettiin uudelleen” COVID-käynteihin. Kaikkien CRF-asiakirjojen poikkeavuuksien tapauskohtainen tarkastelu on myöhemmän analyysin kohteena, mutta seuraavat esimerkit kuvaavat ilmiön vakavuutta ja korostavat tarvetta yksittäisten CRF-asiakirjojen perusteelliseen tarkastukseen. Koehenkilö 12181001 [77], 32-vuotias nainen, ilmoitti ripulista 4. marraskuuta 2020. Maaliskuun 29. päivänä 2021 (16 päivää BLA-hakemuksen jättöajan päättymisen jälkeen ja 3 päivää ennen CRF-asiakirjojen jättöaikaa) tämä haittavaikutus luokitellaan uudelleen ”COVID-käynniksi”, joka on luotu taannehtivasti sponsoreiden ohjeiden mukaisesti.
Koehenkilö 10871286 [78], 71-vuotias nainen, ilmoitti 20. lokakuuta 2020 monenlaisia haittavaikutuksia, jotka tutkimuslääkäri arvioi ”ei vakaviksi” (sivu 648-649). Nämä tapahtumat luokiteltiin uudelleen COVID-oireiksi 29. maaliskuuta 2021.
Lisänäyttöä tietojen manipuloinnista turvallisuustietojen tallenteissa
Toinen merkittävä poikkeama haittavaikutusten kirjaamisessa on huomattava määrä ”kipua pistoskohdissa” toisen lumelääkeannoksen ja ensimmäisen BNT162b2-annoksen määrittämisvaiheen välillä niiden lumelääkkeiden kohdalla, jotka oli sokkoutettu ja joille tarjottiin mRNA-tuotetta. Nämä tapahtumat, jotka on ADAE-tiedostossa merkitty tapahtuneiksi vaiheessa ”Sokkoutuksen purkamisen jälkeen ja ennen rokotusta 3”. Kuten alla olevassa taulukossa esitetään, jotkut koehenkilöt eivät olisi saaneet edes toista annosta [79] — ja vaikka 31:llä (60,8 %) tämä jälkitapahtuma rekisteröitiin samana päivänä kuin heidän kolmas annoksensa, se on oire siitä, että kolmannen annoksen antamisen rekisteröinti viivästyi.
CRF-tiedostojen manuaalisessa tarkastelussa kävi ilmi, että melko usein toimipaikat eivät kirjanneet CRF-tiedostoihin yhtään tai vain osaa esiintyneistä AE:istä. Vain muutamia esimerkkejä:
10551139 [80] — CRF:ssä luetellaan injektiokohdan arkuus ja vilunväristykset, kun taas haittavaikutustaulukossa (ADAE) luetellaan lisäksi vilunväristykset, pahoinvointi, oikean kainalon adenopatia ja injektiokohdan kipu [81].
11401282 [82] — CRF:ssä luetellaan liikunnan aiheuttama astma, kun taas ADAE:ssä luetellaan lisäksi molemminpuolinen keuhkoembolia, syvä laskimotromboosi ja oikean yläkulman vatsakipu sekä poistettu AESPID #2. Koska CRF:ssä on vain AESPID #1, tämä tarkoittaa, että Pfizer lisäsi haittavaikutuksen ja poisti sen ennen kuin lisäsi kolme muuta tapahtumaa, joita ei ole CRF:ssä.
12411347 [83] — CRF:ssä luetellaan päänsärky, injektiokohdan kipu, lymfadenopatia ja toinen tapahtuma päänsärky, kun taas ADAE:ssä on lisäksi kolme tapahtumaa: kuume, päänsärky ja väsymys.
CRF:n AE-loki ei ilmoita yhtään AE:tä 35 koehenkilön osalta [84], ja ainakin 73 CRF:stä puuttuu tietokannassa olevia AE:itä [85], joten tämä poikkeama koskee vähintään 108 koehenkilöä vain 1028 henkilön otoksessa, mikä herättää kysymyksen siitä, onko raportointi tehty yksinomaan sponsoreiden harkinnan mukaan – ja lisäksi siitä, onko CRF-tiedostoja voitu muuttaa. Kyseessä on vähintäänkin vakava GCP-sääntöjen rikkominen, ja jos vahvistetaan, että kyseessä ei ole inhimillinen erehdys, vaan CRF-tiedostoja on muutettu, kyseessä on useiden Yhdysvaltojen [86] ja kansainvälisten säädösten [87] rikkominen.
Koehenkilöiden ilmoittamat COVID-oireet
Jotta BNT162b2:n kykyä ehkäistä COVIDin kaltaisia oireita voitaisiin objektiivisesti kuvata, vaikuttaa tarpeelliselta ottaa huomioon, että jos koehenkilö ilmoitti kuumeesta, olipa Pfizer sitten päättänyt luokitella sen ”haittavaikutuksiksi” tai ”COVID-oireiksi” — tosiasia on edelleen se, että koehenkilö ilmoitti kuumeesta. Kuvasimme haittavaikutukset ja COVID-oireet yhdeksi tiedostoksi [88], joka edusti COVID:n kaltaisia oireita, jotta voisimme edustaa seuraavia ainutlaatuisia uusia tapahtumia, jotka koehenkilöt ilmoittivat ”sokkona” tapahtuneen tarkkailuvaiheen aikana. Katsoimme — protokollan mukaisesti — samoja oireita koskevat ilmoitukset samoista koehenkilöistä neljän päivän sisällä [89] — yhdeksi oireeksi ensimmäisen oireen esiintymispäivänä [90] riippumatta siitä, oliko koehenkilöä koskeneesta tilasta tehty ilmoitus reaktogeenisuustietokannan (FACE), haittatapahtumien (ADAE) vai COVID-oireiden (ADSYMPT) kautta. Tämä tarkoittaa, että jos tutkittava ilmoitti kuumeesta ja yskästä 18. maaliskuuta ja toisesta kuumeesta 19. maaliskuuta, hänet lasketaan vain kerran, 18. maaliskuuta.
Sokkoutuksen purkamista ei satunnaistettu
Plasebo-vastaanottajien sokkouttaminen alkoi heti EUA:n saamisen jälkeen 14. joulukuuta 2020 (FDA antoi luvan siihen 3 päivää EUA:n jälkeen 11. joulukuuta 2020). Koehenkilöiden sokkoutus purettiin ei-sattumanvaraisesti, jolloin iäkkäämmät koehenkilöt asetettiin etusijalle – ja käytännössä lopetettiin satunnaistettu kliininen tutkimus, josta tuli avoin tutkimus, jossa oli hyvin pieni ja erittäin valikoiva plaseboryhmä. Seuraavassa kaaviossa esitetään kumulatiivisten sokkouttamisprosenttien kehitys niiden 44 040 vaiheen 2-3 koehenkilön osalta, jotka olivat saaneet ensimmäisen annoksensa ennen 14. joulukuuta 2020. [91]
Kokeen aikana tapahtuneiden kuolemantapausten oikeuslääketieteellinen analyysi
Vertaisarvioidussa artikkelissa [92] ”Forensic analysis of the 38 subject deaths in the 6-Month Interim Report of the Pfizer/BioNTech BNT162b2 mRNA Vaccine Clinical Trial” (Michels et al., ”Oikeuslääketieteellinen analyysi 38 koehenkilön kuolemantapauksista 6 kuukauden väliraportissa Pfizer/BioNTech BNT162b2 mRNA Vaccine Clinical Trial”) todettiin, että koehenkilö nro 10841470, 65-vuotias lihava latinalaisamerikkalainen mies, jolla oli keuhkofibroosi ja verenpainetauti, kuului Pfizer/BioNTech COVID-19-rokotekokeilun lumelääkekäskyyn. Hän sai annokset 1 ja 2 lumelääkettä 30. syyskuuta ja 21. lokakuuta 2020. Hän sai kuitenkin 23. joulukuuta 2020 annoksen Moderna mRNA -rokotetta, mikä oli tutkimussuunnitelmasta poikkeava toimenpide. Hän sai myöhemmin COVID-19-oireita, joutui sairaalahoitoon ja kuoli lopulta 11. tammikuuta 2021 monielinjärjestelmän vajaatoimintaan. Tapaus ilmoitettiin virheellisesti lumelääkekuolemana, jossa COVID-19 oli toissijainen kuolinsyy, vaikka todellisuudessa koehenkilö olisi pitänyt keskeyttää tutkimuksesta, koska hän oli saanut tutkimukseen kuulumatonta COVID-19-rokotetta.
Asiakirjassa todetaan myös, että ”BNT162b2-rokotetta saaneiden henkilöiden sydäntapahtumat lisääntyivät 3,7-kertaisesti lumelääkkeeseen verrattuna. Niistä 15 koehenkilöstä, jotka olivat aikuisten äkillisiä kuolemantapauksia (SAD) tai löydettiin kuolleina (FD), 12 kuoli sydäntapahtumaan, joista 9 oli rokotettuja.”
Kuolemantapausten viivästynyt kirjaaminen EUA-aikana
Michels et al. korostavat toista merkittävää havaintoa, jota Jeyanthi Kunadhasan kehitti edelleen Australian Therapeutic Goods Administration (TGA) -viranomaisen kanssa käymässään kirjeenvaihdossa. Nämä Australian Medical Professional Societyn (AMPS) puolesta lähetetyt ja julkistetut kirjeenvaihdot koostuvat 21. maaliskuuta 2024 päivätystä ensimmäisestä kirjeestä [93], 27. maaliskuuta 2024 päivätystä TGA:n terveystuotteiden sääntelyryhmän professorin vastauksesta 4.2.2024 [94] ja 17. toukokuuta 2024 [95] päivätystä laajennetusta väitteestä, johon ei ole vielä vastattu. Ne ovat tämän kertomuksen liitteenä. Ne ovat jälleen yksi osoitus sääntelyviranomaisten huolellisuudesta kriittisten sääntöjenvastaisuuksien tutkimisessa — sillä TGA:n keskustelukumppani ei ilmeisesti ymmärrä ensimmäisessä kirjeessä selvitettyjä seikkoja.
Merkittävä poikkeama on kahden kuolemantapauksen viivästynyt ilmoittaminen EUA:n luvuissa, jotka ilmoitettiin sääntelyviranomaisille ja maailmalle joulukuussa julkaistun Polackin ja muiden tutkijoiden NEJM-tutkimuksen [96] kautta. Polackin julkaisussa ilmoitettiin kuusi kuolemantapausta – kaksi BNT162b2-haarassa ja neljä lumelääkehaarassa. Sekä lehtiartikkelissa että EUA-hyväksyntäasiakirjoissa todettiin kuusi kuolemantapausta 27. heinäkuuta 2020 ja 14. marraskuuta 2020 välisenä aikana. Artikkelista käy ilmi, että Pfizer-BioNTechin kirjanpidossa oli kahdeksan kuolemantapausta EUA-aikana tapahtuneista 11 kuolemantapauksesta. Kirjoittajat osoittavat, että Pfizerilla oli selkeä tieto neljästä BNT162b2-haarassa ja neljästä lumelääkehaarassa, ja että Pfizerin olisi pitänyt ilmoittaa näistä FDA:lle. Kaksi julkistamatonta kuolemantapausta osoitti sydäntapahtumasignaalin kliinisen tutkimuksen BNT162b2-hoitoa saaneilla henkilöillä. Koehenkilö 11141050 kuoli 19. lokakuuta 2020, eli hyvissä ajoin ennen tietojen päättymispäivää 14. marraskuuta 2020. Asiakirjoista käy ilmi, että koehenkilön hätäyhteyshenkilö ilmoitti kuolemasta kliiniselle tutkimuspaikalle kuolinpäivänä, kuten protokollan vaatimukset edellyttävät. Tutkimussuunnitelmassa edellytettiin myös, että kliininen tutkimuspaikka ilmoittaa Pfizerille rokotteen SAE-lomakkeella 24 tunnin kuluessa kuolemantapauksen ilmoittamisesta. Kliininen henkilökunta odotti kuitenkin 37 päivää, ennen kuin potilaan kuolema kirjattiin Pfizerin tietoihin. Viivästyksen vuoksi Pfizer ei toimittanut tätä kuolemantapausta osana EUA:n tietoja, mikä herättää kysymyksiä viivästyksen syistä ja mahdollisista hyvän kliinisen hoitokäytännön rikkomisista.
Ruumiinavauksen mukaan potilas kuoli ”äkilliseen sydänkuolemaan”, ja hänen tiedossaan olevat riskitekijät, kuten korkea verenpaine ja liikalihavuus, aiheuttivat hänelle suuren riskin saada äkillinen sydäninfarkti. Kliinisen hoitopaikan henkilökunta kirjasi potilaan muistiinpanoihin erityisen diagnoosin ”äkillinen sydänkuolema” 9. joulukuuta 2020, päivää ennen rokotteiden ja niihin liittyvien biologisten tuotteiden neuvoa-antavan komitean (VRBPAC) kokousta 10. joulukuuta 2020, mikä viittaa siihen, että myös tämän salatun kuolemantapauksen ruumiinavaustulokset olivat saatavilla rokotteen hätäkäyttöluvan kriittisessä käsittelyvaiheessa.
Osallistuakseen tähän kliiniseen tutkimukseen osallistujien oli oltava terveitä anamneesin, fyysisen tutkimuksen (jos tarpeen) ja tutkijan kliinisen arvion perusteella. Tutkimussuunnitelmassa sallittiin terveiden osallistujien osallistuminen kliiniseen tutkimukseen, joilla oli jo ennestään vakaa sairaus, joka määriteltiin sairaudeksi, joka ei vaatinut merkittävää hoitomuutosta tai sairaalahoitoa sairauden pahenemisen vuoksi kuuden viikon aikana ennen tutkimukseen osallistumista. Verenpainelukema puuttuu hänen julkisesti saatavilla olevista potilaskertomuksistaan. Näin ollen voimme vain olettaa, että potilaan korkea verenpaine, josta hän oli kärsinyt 1. tammikuuta 2010 lähtien, oli hyvin hallinnassa, kun hänet otettiin tutkimukseen. Potilas painoi 74,1 kg ja oli 165 cm pitkä. Näin ollen hänen painoindeksinsä 27,2 sijoitti hänet ylipainoluokkaan, ei lihavaan luokkaan, kuten kohdassa 74 todetaan.
Koehenkilö 11201050 kuoli 7. marraskuuta 2020. Hänen aviomiehensä ilmoitti hänen kuolemastaan kliiniselle tutkimuspaikalle 7. marraskuuta 2020. Hän kuoli unissaan 72 päivää sen jälkeen, kun hän oli saanut rokoteannoksen 2. Sairaalakäyntiä tai ruumiinavausta ei tehty. Kuolemansyyntutkija totesi hänen kuolemansa ja merkitsi kuolintodistukseen kuolinsyyksi sydänpysähdyksen.
Koska ruumiinavaustuloksia ei ollut saatavilla, on epäselvää, miten rahoittajat tai sääntelyviranomaiset päättelivät, että kuolemantapauksen ei voitu katsoa johtuneen hoidosta. Pfizer dokumentoi saaneensa ilmoituksen hänen kuolemastaan 7. marraskuuta 2020, paljon ennen tietojen päättymispäivää 14. marraskuuta 2020. On selvitettävä, miksi tätä rokotukseen liittyvää kuolemantapausta ei julkistettu VRBPAC:n kokouksessa 10. joulukuuta 2020 tai Polack New England Journal of Medicine -julkaisussa.
Vahvistetaan Michelsin ym. arvio massiivisesti aliraportoiduista kuolemantapauksista
Michels et al. toteavat, että ”koehenkilöiden kuolemantapausten määrä oli 17 % odotetusta määrästä, joka perustui ikään mukautettuun kuolleisuuteen Yhdysvalloissa. Yksi mahdollinen selitys voisi olla 395 koehenkilön ”jatkotutkimuksista katoaminen”. Toinen selitys näille alhaisille kuolemantapauksille voisi olla se, että tutkijoiden tiedostoissa oli 1203 tutkittavaa, jotka on merkitty kohdissa 23-25 – ja/tai se, että kohdissa 47-51 on merkitty (ainakin) 301 puuttuvaa tietuetta.
Yksikään BNT162b2-vastaanottaja ei kuollut raportoitujen lukujen mukaan Yhdysvaltojen ulkopuolella. Kuolemantapaukset Yhdysvalloissa ovat 19 BNT162b2:n jälkeen, 12 plasebon jälkeen ja 2 plaseboa ensin saanutta henkilöä, jotka olivat sen jälkeen saaneet vähintään yhden BNT162b2-annoksen, eli yhteensä 33 kuolemantapausta Yhdysvalloissa. Viisi lumelääkettä saanutta henkilöä kuoli Yhdysvaltojen ulkopuolella.
Otimme kuolleisuustiedot osavaltioittain, vuoden, kuukauden ja 5 vuoden ikäryhmien mukaan vuosilta 2020 ja 2021 CDC:n Wonder Underlying Causes of Death -alustalta [97] — ja loimme vientitiedot [98] sekä toisen vientitiedon [99] väestötiedoista [100], jotka on kerätty vuosittain heinäkuun 1. päivän väestölaskennan tietojen perusteella osavaltioittain. Tämän avulla voitiin vertailla Yhdysvaltojen tutkimuslaitosten raportoimaa kuolleisuutta ( ja kuolleisuutta, jota olisi voitu odottaa tämän ikäiseltä kohortilta. Terveiden käyttäjien harhapainotteisuus voisi mahdollisesti vaikuttaa asiaan, mutta se, että havaittiin yksi ”dementiaan” johtanut kuolema, antaa aiheen kyseenalaistaa tutkimuksen aikana rekrytointiin sovelletut standardit. Tämä ei kuitenkaan voi selittää sitä, miten merkittävästi pienempi havaittu kuolleisuus on kuin mitä olisi voitu odottaa 33689 satunnaistetun henkilön [101] populaatiossa — vain 18 prosenttia odotetusta kuolleisuudesta on raportoitu.
Prosessi 1 & prosessi 2: yleiskatsaus niiden eroista
Pfizer/BioNTech käytti COVID-19-rokotekandidaatin BNT162b2:n (kauppanimeltään Comirnaty) kehityksessä kahta erillistä valmistusmenetelmää, joita he kutsuivat ”prosessiksi 1” ja ”prosessiksi 2”.
Kliinisen tutkimuksen tutkimussuunnitelmassa [102] todetaan: ”Alkuperäinen BNT162b2 valmistettiin ’prosessilla 1’, mutta ’prosessi 2’ kehitettiin tukemaan valmistuksen laajempaa mittakaavaa.” Lisänäyttöä löytyy muista Yhdysvaltain FDA:n, Japanin lääke- ja lääkinnällisten laitteiden viraston (PMDA), TGA:n ja Euroopan lääkeviraston (EMA) sääntelyasiakirjoista. Yhtenä esimerkkinä mainittakoon, että EMA:n 19. helmikuuta 2021 päivätyssä Comirnatya koskevassa tuotearviointikertomuksessa (PAR) todetaan seuraavaa: ”Kehityshistorian aikana on käytetty kahta vaikuttavan aineen prosessia: prosessi 1 (kliinisen tutkimuksen materiaali) ja prosessi 2 (kaupallinen prosessi) ” [103].
Kohdassa 83 mainitun EMA:n PAR:n lausunnon lisäksi, jonka mukaan prosessia 1 käytettiin kliinisten koeannosten valmistukseen ja prosessia 2 kaupallisiin annoksiin, Japanin PMDA:n 8. helmikuuta 2021 päivätyssä ”Report on Special Approval for Emergency” (raportti hätätilanteessa myönnettävästä erityishyväksynnästä) lausunnossa todetaan seuraavaa: ”Ei-kliinisissä ja kliinisissä tutkimuksissa käytetty vaikuttava aine valmistetaan prosessilla 1 ja ehdotetun kaupallisen formulaation vaikuttava aine prosessilla 2” [104].
Lokakuun 6. [105] päivättyyn pöytäkirjaan tehdyssä tarkistuksessa 7 todetaan seuraavaa: ”Lisättiin ylimääräinen tutkiva tavoite turvallisuuden ja immunogeenisuuden kuvaamiseksi 16-55-vuotiailla osallistujilla, jotka on rokotettu tutkimusinterventiolla, joka on tuotettu valmistusmenetelmällä ’Process 1’ tai ’Process 2’.”. Tätä muutosta käsitellään tarkemmin kohdassa 6.1.1: ”BNT162b2:n valmistuksen mittakaavaa on kasvatettu tulevien toimitusten tukemiseksi. BNT162b2:ta, joka on tuotettu lisääntynyttä tarjontaa tukevalla valmistusprosessilla (”prosessi 2”), annetaan tutkimuksessa noin 250:lle 16-55-vuotiaalle osallistujalle erää kohti. Tutkimuksessa kuvataan ennaltaehkäisevän BNT162b2:n turvallisuutta ja immunogeenisuutta 16-55-vuotiailla henkilöillä, jotka on rokotettu materiaalilla, joka on tuotettu nykyisellä valmistusprosessilla (prosessi 1), ja materiaalilla, joka on tuotettu eristä, jotka on tuotettu lisääntynyttä tarjontaa tukevalla valmistusprosessilla (prosessi 2) ” [106].
Kohdassa 65 siteeratussa kliinisen tutkimuksen pöytäkirjassa esitetään tiivistetysti kolme keskeistä eroa prosessien 1 ja 2 välillä: ”Prosessimuutokset liittyvät DNA-mallin tuotantomenetelmään, josta RNA-lääkeaine transkriboidaan, ja RNA-lääkeaineen puhdistusmenetelmään. BNT162b2-lääkevalmistetta valmistetaan tämän jälkeen käyttämällä skaalattua LNP-valmistusprosessia ” [107].
Erot prosessien 1 ja 2 välillä eivät ole vähäpätöisiä. TGA:lta saadussa asiakirjassa [108] esitetään yksityiskohtaisesti joitakin keskeisiä muutoksia, jotka on tehty prosessien 1 ja 2 välillä, vaikka monet yksityiskohdat on poistettu. Prosessissa 1 mRNA:n transkriptioon tarvittava DNA-malli tuotettiin PCR-amplifikaation avulla; prosessissa 2 käytetään linearisoitua plasmidi-DNA:ta, jota viljellään E. coli -bakteereissa. Linearisoidussa plasmidi-DNA:ssa käytetään plasmidi-DNA:ta, joka on pieni, pyöreä, kaksijuosteinen DNA-molekyyli, jota tavallisesti esiintyy bakteereissa. Muissa (osittain redusoiduissa) yksityiskohdissa kuvattiin muutoksia in vitro -transkriptioreaktion tilavuuteen, erän mittakaavaan ja puhdistusmenetelmään (magneettihelmet prosessissa 1; proteinaasi K -käsittely proteiinien pilkkomiseksi ja niihin liittyvien nukleiinihappojen vapauttamiseksi, jota seuraa tangentiaalinen virtaussuodatus prosessissa 2).
Prosessin 2 erien tunnistaminen
Joissakin sääntelyasiakirjoissa prosessiin 1 viitataan joskus ”klassisena” prosessina ja tuloksena olevaan tuotteeseen viitataan ”kliinisenä toimituksena”. Prosessiin 2 viitataan toisinaan nimellä ”upscale”-prosessi, ja sen tuloksena syntyviin tuotteisiin viitataan nimellä ”emergency” tai ”commercial” supply. Tämä käy ilmi TGA:n julkaisemasta asiakirjasta. Kyseessä on FOI 3659 -asiakirja 4, jonka otsikko on ”BNT162b2 (PF-07302048) Comparability Report for PPQ Drug Product Lots ”109 ja jossa luetellaan jaetut erät, niiden käyttö ja vastaavat tuotantoprosessit.
Vertailukelpoisuusraportissa erotellaan kliiniset, hätä- ja kaupalliset toimituserät seuraavasti: ”Lääkevalmisteen valmistusprosessi on kehittynyt kliinisestä toimituksesta hätäaputoimituksiin ja lopulta kaupallisiin toimituksiin, jolloin se on siirretty eri valmistuspaikkoihin ja prosessia on skaalattu. Kliininen tuotanto valmistettiin alun perin Polymunissa Itävallassa (”klassinen” prosessi). LNP-prosessin massan läpimenon parantamiseksi ja eräkoon kasvattamiseksi prosessi skaalattiin Polymunissa (”upscale”-prosessi), minkä jälkeen irtotavarana oleva lääkevalmiste (valmiiksi formuloidut LNP:t ennen steriilisuodatusta) kuljetettiin Pfizer Puursiin, Belgiaan täyttöä/viimeistelyä varten (hätäaputoimitusten ja kaupallisten toimitusten valmistusta varten). Tämä prosessi on otettu käyttöön myös mibe:ssä (Dermapharm) Saksassa hätäaputarvikkeiden valmistusta varten. Tässä yhteydessä Puursin täyttö-/viimeistelytoiminnot suoritettiin aluksi S2F2-linjalla (kliinisiä toimituksia varten) ja siirryttiin WSL5-linjalle suurempien erämäärien valmistamiseksi (hätätoimitukset ja myöhemmin kaupalliset toimitukset). Rutiininomaista kaupallista tuotantoa varten LNP-tuotantoprosessi on siirretty kokonaan Pfizeriin, Puursiin, Belgiaan, jossa täyttö-/viimeistelytoiminnot tapahtuvat täyttölinjoilla WSL5, FC2 ja VC2, ja Pfizeriin, Kalamazoon, Yhdysvaltoihin, jossa täyttö-/viimeistelytoiminnot tapahtuvat täyttölinjoilla L8 ja L18 (kaupallisten toimitusten valmistusta varten).” [110].
Samasta asiakirjasta voidaan tunnistaa, mitkä erät ovat prosessia 1 tai prosessia 2, sivun 4 taulukossa 1 ja sivun 11 taulukon 3 huomautuksissa b ja e lueteltujen erien perusteella. Lisätietoa erien valmistusprosessista löytyy FDA:n FOI:n perusteella julkaisemasta asiakirjasta [111]. Asiakirjan nimi on ”125742_S2_M3_32p5_batch_analyses.pdf”. – Asiakirjan sivulla 4 oleva taulukko 3.2.P.5.4-1 ”Summary of BNT162b2 Drug Product Lots” (Yhteenveto BNT162b2-lääke-eristä) sisältää asiaa koskevat tiedot. Jäljempänä viitataan molempiin asiakirjoihin.
Kliinisessä lääketutkimuksessa C4591001 käytettyjen prosessin 2 erien määrittäminen
FDA:n julkaisemassa asiakirjassa kerrotaan yksityiskohtaisesti, mitkä erät oli toimitettu millekin kliiniselle tutkimuspaikalle noin 6 kuukauden kuluttua tutkimuksen alkamisesta. Se on päivätty 17. maaliskuuta 2021 ja otsikoitu ”125742_S1_M5_5351_c4591001-interim-mth6-patient-batches.pdf ” [112]. Asiakirjassa luetellaan seuraavat yksilölliset eränumerot: BCV10320-A, BCV10420-A, BCV20420-A, BCV40420-A, BCV40620-A, BCV40620-B, BCV40620-C, BCV40620-D, BCV40720-A, BCV40720-B, BCV40720-C, ED3938, EE3813, EE8493Z ja EJ0553Z. Ei ole selvää, mitä erien EE8493 ja EJ0553 lopussa oleva Z tarkoittaa. Muissa asiakirjoissa nämä erät on lueteltu ilman Z-liitettä. Sitä on saatettu käyttää osoittamaan, että nämä kokeessa käytetyt erät olivat peräisin prosessista 2.
Edellä 88 ja 90 kohdassa mainittujen TGA:n ja FDA:n asiakirjojen taulukoiden perusteella kaikki erät, jotka alkavat kirjaimella ”BCV”, ovat prosessin 1 eriä. Erä ED3983 on lueteltu 90 kohdassa mainitun FDA:n eränanalyysiasiakirjan taulukossa 3.2.P.5.4-1 ”kliinisenä” eränä, ja se on merkitty huomautuksella a, joka on määritelty sivulla 13 seuraavasti: ”vastaa BCV40720-P:tä”, joka vahvistettiin prosessin 1 eräksi 91 kohdassa. Erä EE3813 on molemmissa taulukoissa merkitty prosessin 1 kliiniseksi eräksi.
Kohdassa 91 mainitussa 6 kuukauden potilaseriä koskevassa asiakirjassa yksilöidään kaksi prosessin 2 erää: EE8493 ja EJ0553. Erä EE8493 mainitaan prosessin 2 eränä edellä 90 kohdassa mainitun TGA:n ”Comparability Report for PPQ Drug Product Lots” -asiakirjan taulukossa 1 (ks. sarake ”DS Process and Site of Manufacture”). Erä EJ0553 on lueteltu TGA:n vertailukelpoisuusraportissa sivulla 11 olevan taulukon 3 huomautuksessa ”d”. Taulukon 3 huomautus ”d” on liitetty sarakkeeseen ”Historical Range: Emergency Supply”, mikä osoittaa, että EJ0553 on hätäaputoimituserä, joka oli toinen termi prosessille 2. Huomautuksessa ”d” täsmennetään lisäksi, että erän EJ0553 valmistuspaikka oli ”Polymun/Puurs WSL5”, mikä kohdassa 88 siteeratun asiakirjan kuvauksen perusteella antaa lisätodisteita siitä, että EJ0553 oli hätäaputoimituserä eikä kliininen erä, koska kaikki taulukossa 1 olevat erät, joiden ”DP Site of Manufacture” -kenttään on merkitty ”Polymun/Puurs WSL5”, ovat prosessi 2 -eriä. Tämä on täysin yhdenmukainen sivulla 3 olevan valmistusprosessin kehittymistä koskevan kuvauksen kanssa, jota on lainattu edellä 70 kohdassa. Monet muut huomautuksessa ”d” mainitut erät on lueteltu TGA:n vertailukelpoisuusraportin sivuilla 4-5 olevassa taulukossa 1, ja ne on kaikki merkitty prosessin 2 eriksi, eikä yksikään huomautuksessa ”d” taulukossa 3 luetelluista eristä ole taulukossa 1 kliinisenä eränä (prosessi 1). Lisäksi kaikki kliiniset erät on lueteltu erikseen taulukon 3 huomautuksessa ”b” sivulla 11. Erä EJ0553 on lueteltu kohdassa 90 mainitussa FDA:n ”Batch Analysis” -asiakirjassa sekä hätätoimitus- että kliinisenä eränä. Koska erä EE8493 on myös lueteltu sekä hätätoimituseränä että kliinisenä eränä, tämä osoittaa, että sekä EE8493:aa että EJ0553:a käytettiin jossakin määrin kliinisessä tutkimuksessa ja että ne olivat myös osa hätätoimituserää.
Lisäksi lähes kaikki haittavaikutukset, jotka on raportoitu [113] Yhdysvaltojen tautienvalvontakeskuksen rokotteiden haittavaikutusilmoitusjärjestelmässä (VAERS [114]) erien EE8493 ja EJ0553 osalta, on rokotettu sen jälkeen, kun Comirnaty oli hyväksytty väliaikaiseen käyttöön Yhdistyneessä kuningaskunnassa. VAERS-järjestelmässä [115] ei ole raportteja haittavaikutuksista yhdestäkään muusta erästä, joka on lueteltu tutkimuspaikoille toimitetuksi. Tämä antaa lisätodisteita siitä, että EE8493 ja EJ0553 olivat prosessin 2 valmisteita, koska niitä jaettiin ja annettiin kaupallisesti myyntiluvan myöntämisen jälkeen.
Todisteet siitä, että erää EE8493 käytettiin prosessien 1 ja 2 suunniteltuun vertailuun
On kaksi tapaa tunnistaa, että erää EE8493 käytettiin osana suunniteltua vertailututkimusta prosessien 1 ja 2 välillä, kuten 85 kohdassa mainitussa kliinisen tutkimuksen pöytäkirjassa on kuvattu. Ensimmäinen tapa on tunnistaa, mitkä prosessin 2 erät toimitettiin mihin kliinisen tutkimuksen tutkimuspaikkoihin, ja laskea, kuinka monta hoidettavaa otettiin mukaan vertailututkimuksen alusta alkaen (ks. jäljempänä kohdat 98-99). Toiseksi osoitetaan, että osallistujien osajoukon satunnaistamisnumerot erosivat kaikista muista (ks. jäljempänä kohta 100). Nämä toisistaan riippumattomat menetelmät johtavat samaan tulokseen: vain 252 koehenkilöä, joille annettiin rokoteannoksia erästä EE8493, otettiin lopulta mukaan suunniteltuun vertailuun.
Vastauksessaan FOI-pyyntöön 23/510 Yhdistyneen kuningaskunnan Medicines & Healthcare products Regulatory Agency (MHRA) todisti, että ”ensimmäinen kliininen erä, joka sisälsi prosessin 2 lääkeaineita, annosteltiin 19. lokakuuta 2020 Yhdysvalloissa ” [116].
Kohdassa 91 mainitussa FDA:n asiakirjassa luetellaan, mitkä erät oli jaettu mille tutkimuspaikoille 17. maaliskuuta 2021 mennessä. Samankaltaisessa asiakirjassa [117] luetellaan, mitkä erät oli toimitettu kullekin tutkimuspaikalle 19. marraskuuta 2020 mennessä. Kyseisessä asiakirjassa ainoa Process 2 -erä, joka on lueteltu, on EE8493 (huomautus: erää EJ0553 ei ole lueteltu). Näin ollen voidaan päätellä, että EE8493 oli ainoa prosessin 2 erä, joka oli jaettu kliinisten tutkimusten tutkimuspaikoille, kun prosessien 1 ja 2 vertailututkimus alkoi 19. lokakuuta 2020. Sitä toimitettiin tutkimuspaikkoihin 1133, 1135, 1146 ja 1170 sekä välivaiheen että 6 kuukauden potilaseräasiakirjojen mukaan.
Niiden koehenkilöiden analyysi, jotka saivat ensimmäisen annoksensa 19. lokakuuta 2020 tai sen jälkeen [118], osoittaa, että näissä neljässä toimipisteessä oli yhteensä 252 koehenkilöä, jotka saivat rokotteen, ja 250 koehenkilöä, jotka saivat lumelääkettä [119]. Kaikki hoitohaaran 252 koehenkilöä olivat 16-55-vuotiaita. Tämä vastaa tutkimussuunnitelmassa esitettyä kuvausta suunnitellusta tutkimuksesta, jonka mukaan Process 2 -annoksia annettaisiin ”noin 250:lle 16-55-vuotiaalle osallistujalle”, kuten kohdassa 2 on yksityiskohtaisesti kuvattu. (Ks. liite A, jossa on luettelo 252 koehenkilöstä, jotka rekrytoitiin kyseisiin toimipaikkoihin kyseisenä ajanjaksona). Tämä analyysi voidaan vahvistaa tarkastelemalla silmämääräisesti BLA:n satunnaistamistiedostoa, jonka toimeksiantajat ovat toimittaneet FDA:lle [120].
Erä EJ0553 toimitettiin toimipaikkaan 1007. Kohteessa 1007 19. lokakuuta 2020 jälkeen hoidon saaneilla 17:llä vastaanottajalla ei ole satunnaistamisnumeroita, jotka osoittavat, että heidät on sisällytetty Prosessien 1 ja 2 vertailututkimukseen, kuten kohdassa 100 on esitetty. Lisäksi vain 17 hoidettavaa henkilöä olisi voinut mahdollisesti saada annoksia erästä EJ0553, eikä noin 250:tä, kuten kohdassa 2 mainitussa pöytäkirjassa on suunniteltu. Näin ollen voimme päätellä, että nämä 17 koehenkilöä eivät kuuluneet suunniteltuun tutkimukseen, jossa verrattiin prosesseja 1 ja 2. Kun otetaan huomioon, että erä EJ0553 toimitettiin kolmelle muulle tutkimuspaikalle, joihin ei 14. marraskuuta 2020 jälkeen otettu uusia hoitohenkilöitä, on todennäköistä, että annokset tästä prosessin 2 erästä toimitettiin näille neljälle tutkimuspaikalle annettavaksi lumelääkehenkilöille, jotka vapautettiin sokkouttamatta ja jotka tarjoutuivat rokotettaviksi sen jälkeen, kun FDA oli myöntänyt kiireellisen käyttöluvan 11. joulukuuta 2020.
Tässä analyysissä käytetyistä aineistoista ja asiakirjoista löytyy toinen keino vertailututkimukseen osallistuneiden koehenkilöiden tunnistamiseksi, jonka löysivät samat kirjoittajat kuin Michels et al. (mainittu kohdissa 71-79): koehenkilön satunnaistamisnumero [121]. Jokainen tutkimukseen osallistunut koehenkilö sijoittuu numeroiden 1081 ja 274318 väliin, lukuun ottamatta pientä 502 koehenkilön osajoukkoa, jonka satunnaistamisnumerot ovat välillä 400002 ja 401509. Näistä 252 on samoja hoitohenkilöitä, jotka on tunnistettu kohdassa 98 neljässä toimipaikassa, jotka saivat Process 2 -eriä ennen 19. marraskuuta 2020. Loput 250 ovat lumelääkettä saaneita, jotka ovat samoja kuin kohdassa 97 [122] yksilöidyt neljä toimipaikkaa. Tämä vahvistaa riippumattomasti kohdassa 80 esitetyn päätelmän, jonka mukaan nämä 252 hoitoa saanutta koehenkilöä, jotka olivat peräisin erän EE8493 saaneista toimipaikoista, olivat ainoat, jotka oli tarkoitettu prosessien 1 ja 2 väliseen vertailuun.
Prosessin 1 ja prosessin 2 mukaisen koehenkilöiden värväyksen vertailu on esitetty viikkokaaviossa.
Todisteet siitä että Prosessien 1 ja 2 suunniteltua vertailua ei koskaan suoritettu
Vastauksessaan FOI-pyyntöön 23/510, johon viitataan kohdassa 96 ja joka on tämän kertomuksen liitteenä, MHRA toteaa, että tutkimustuloksia odotettiin helmikuussa 2021 (sivu 2). Suunniteltu vertailu pöytäkirjasta kuitenkin ”poistettiin ja dokumentoitiin pöytäkirjamuutoksella 20 syyskuussa 2022, koska ’prosessin 2’ kautta valmistettuja rokotteita käytettiin laajasti.”. Näin ollen tätä prosessivertailua ei suoritettu osana virallista dokumentointia” (sivu 5). Ainakaan MHRA:n mielestä vertailututkimusta ei koskaan suoritettu.
Todisteet siitä, että nämä kaksi tuotetta eivät ole täysin samanlaisia: Prosessi 2 -valmisteen saajien keskuudessa esiintyy enemmän haittavaikutuksia
Mallinnimme prosessin 2 vastaanottajana olemisen vaikutusta muihin muuttujiin verrattuna yhdysvaltalaisten, 55-vuotiaiden tai sitä nuorempien koehenkilöiden rekisteröimien haittavaikutusten määrään Poissonin regression avulla ja havaitsimme, että se lisää haittavaikutusten lukumäärää 264,33 prosentilla; sillä on erittäin merkittävä vaikutus haittavaikutuksiin (6,59 kertaa suurempi vaikutus kuin ”liitännäissairauksilla”), vaikkakin se on vähemmän merkittävä kuin sukupuolen tai liikalihavuuden vaikutus [123].
Verrattaessa yksinomaan 95:tä ei-lihavuutta sairastavaa miespuolista vastaanottajaa Yhdysvalloissa prosessi 2 johtaa 211,77 prosentin lisäykseen, jonka vaikutus on 4,84 kertaa suurempi kuin komorbiditeetin vaikutus. Arviointi on samanlainen (214,49 % lisäys) negatiivisen binomiregression avulla.
Todisteet eroista lymfadenopatiassa prosessin 1 ja prosessin 2 erien välillä ja MHRA:n epätarkat lausunnot
Lymfadenopatia on lääketieteellinen termi turvonneille imusolmukkeille. Kliinisen tutkimuksen (C4591001) kahden ensimmäisen rokoteannoksen ja kolmannen (tehosterokote) rokoteannoksen aiheuttaman lymfadenopatian esiintyvyydessä havaitut erot viittaavat prosessin 1 ja prosessin 2 rokotevalmisteen erilaisiin haittavaikutusprofiileihin. Syynä on se, että kliinisen tutkimuksen kaksi ensimmäistä annosta olivat valtaosin prosessilla 1 valmistettuja rokoteannoksia, kun taas useita kuukausia myöhemmin annetut tehosteannokset olisi valmistettu prosessilla 2 kaupallista toimitusta varten.
Pfizerin/BioNTechin COVID-19 mRNA-rokotteen BNT162b2 väliaikaista toimituslupaa koskevassa ”MHRA’s Public Assessment Report for the Authorisation for Temporary Supply” -raportissa (MHRA:n julkinen arviointiraportti) kuvataan lymfadenopatian määrää kliinisen tutkimuksen C4591001124 alustavan raportin perusteella. Vaikka julkinen arviointiraportti (MHRA:n PAR) päivitettiin nuorten rokotusten osalta kesäkuussa 2021, kansilehdellä todetaan, että siinä ”esitetään yhteenveto alkuperäisestä arviosta hyväksynnän myöntämishetkellä joulukuussa 2020.”. Alkuperäisen raportin teksti säilyy ennallaan”.
Kliinisen tutkimuksen C4591001 lymfadenopatian määrä on MHRA:n PAR:n sivulla 42 lueteltu 0,3 prosentiksi ja luokiteltu sivulla 50 ”harvinaiseksi”, joka on määritelty esiintyväksi yli 0,1 prosentissa tapauksista mutta alle 1 prosentissa tapauksista.
MHRA:n kesäkuussa 2023 julkaistussa asiakirjassa ”Information for Healthcare Professionals on COVID-19 Vaccine Pfizer/BioNTech (Regulation 174)” lymfadenopatian määrä on esitetty taulukossa 1 [125]. Se on lueteltu harvinaiseksi. Mutta taulukon alaosassa olevassa huomautuksessa (a) todetaan: ”Lymfadenopatian esiintyvyys oli suurempi (5,2 % vs. 0,4 %) osallistujilla, jotka saivat tehosteannoksen (kolmas annos), verrattuna osallistujiin, jotka saivat kaksi annosta.”” Lymfadenopatian 5,2 %:n esiintymistiheys siirtäisi lymfadenopatian luokasta ”harvinainen” luokitukseen ”yleinen”. Se merkitsee yli 13-kertaista kasvua lymfadenopatian määrässä [126].
Yksi mahdollinen selitys tälle lisääntymiselle on se, että tehosteannos saa aikaan voimakkaamman immuunireaktion, mikä johtaa lymfadenopatian lisääntymiseen. Tel Avivin yliopiston lääketieteellisen tiedekunnan israelilaistutkijoiden PET-CT-kuvauksia käyttäen suorittamissa huolellisissa ja erittäin tarkoissa lymfadenopatiaa koskevissa kuvantamistutkimuksissa todettiin kuitenkin, että ”minkä tahansa asteisen VAHL:n [rokotteeseen liittyvä hypermetabolinen lymfadenopatia] kokonaisesiintyvyys kolmannen COVID-19-rokoteannoksen jälkeen on pohjimmiltaan samankaltainen kuin mitä raportoitiin ensimmäisten ja toisten COVID-19-rokoteannosten jälkeen. Kolmannen annoksen jälkeisten VAHL-tapausten havaittiin kuitenkin olevan lyhyempikestoisia ja niiden tartuntaintensiteetti oli alhainen viiden ensimmäisen päivän kuluttua rokotuksesta ” [127]. Tämä tarkoittaa, että tehosterokotuksen jälkeinen lymfadenopatia ei ollut pahempi kuin kahden ensimmäisen rokoteannoksen jälkeen, vaan se oli joiltakin osin jopa lievempi.
Israelilaisen tutkimuksen tulokset eivät ole yhdenmukaisia MHRA:n ilmoittaman lymfadenopatian 13-kertaisen lisääntymisen kanssa kahden ensimmäisen annoksen ja kolmannen annoksen välillä. Tätä israelilaista tutkimusta ei tehty osana kliinistä tutkimusta C4591001. Se perustui henkilöihin, jotka saivat kaupallisia annoksia, jotka toimitettiin Israeliin rokotteen käyttöönoton jälkeen, eli annoksia, jotka oli valmistettu prosessilla 2.
Tämän perusteella voidaan erittäin suurella varmuudella päätellä, että MHRA:n ilmoittama lymfadenopatian lisääntyminen johtuu siirtymisestä kliinisessä tutkimuksessa käytetyistä prosessin 1 annoksista prosessin 2 annoksiin, joita käytettiin myöhemmin tehosteannoksina.
Vaikka lymfadenopatian 5,2 prosentin osuuden lähdettä ei ole ilmoitettu, kun otetaan huomioon ajankohta, jolloin tehosteannokset annettiin ensimmäisen kerran vuoden 2021 jälkipuoliskolla, niiden on täytynyt olla peräisin prosessia 2 käyttäen annetuista tehosteannoksista. Lymfadenopatian lisääntynyttä määrää tehosteannoksissa tukee myös Pfizer/BioNTechin kliininen tutkimus C4591031, joka koski ensimmäistä tehosteannosta (kolmas annos) ja jossa lymfadenopatian osuus oli 2,7 % [128]. Tämä merkitsee yli kuusinkertaista kasvua annoksiin 1 ja 2 verrattuna, eikä se myöskään ole yhdenmukainen edellä mainitun israelilaisen tutkimuksen tuloksen kanssa (ks. viite 12), jonka mukaan lymfadenopatian määrässä ei ole eroa ensimmäisen tehosteannoksen ja aikaisempien annosten välillä.
Katkaisupäivät eivät olleet “tietojen katkaisupäiviä”
Perinteisissä tutkimuksissa on rajapyykki, joka vastaa tutkimuksen kronologista päättymispäivää, mutta tässä tapauksessa tutkimus jatkui sekä EU:n myyntiluvan että BLA-hakemuksen jättämisen jälkeen. Käytännössä, vaikka BLA-aineistossa esitettiin asiakirjoja, joiden mukaan tutkimuksen päättymispäivä oli 13. maaliskuuta 2021, CRF-tiedotteita muokattiin aina 1. huhtikuuta 2021 asti, kuten minkä tahansa CRF-tiedoston silmämääräinen tarkastelu osoittaa — ks. esim. kohta 65. Tämä tekee BLA-hakemuksen jäljentämisen — kuten ilmoitettujen COVID-tapausten tarkan jäljentämisen — lähes mahdottomaksi ilman täydellistä pääsyä kirjausketjuun. Valtaosa ”COVID-oireiksi uudelleen kelpuutetuista” haittavaikutuksista liittyy näihin ”viime hetken muokkauksiin”. Esimerkkinä voidaan mainita vain muutamia 1028 CRF-tiedostosta löytyviä tietojen rajaamiseen liittyviä epäselvyyksiä:
12231058 [129] — 2. ja 3. maaliskuuta 2021 ilmenneet väsymystä ja pistoskohdan kipua koskevat haittavaikutukset kirjattiin 23. maaliskuuta 2021, ja ne ovat tietokannassa [130].
12261769 [131] — Koehenkilöllä on COVID-käynti 18. maaliskuuta 2021, joka on merkitty 19. maaliskuuta 2021 ja jota ei ole tietokannassa.
12312420 [132] — COVID-käynti A 12.3.-15.3.2021, merkitty 16.3.2021, ja COVID-käynti B, 26.3. – jatkuva, merkitty 29.3.2021, jossa positiivinen paikallinen pyyhkäisynäyte sivulla 235, eivät molemmat ole tietokannassa.
12313610 [133] — 19. helmikuuta 2021. tapahtunut eteislepatuksen SAE kirjattiin 20. maaliskuuta 2021, kun koepaikka oli lähettänyt alkuperäisen turvallisuustietokannan 19. maaliskuuta 2021, sivu 181, ja se on tietokannassa.
Lääketieteellisen historian porsaanreiän hyväksikäyttö
Tutkimuspöytäkirjassa [134] määriteltiin merkittäviä porsaanreikiä haittatapahtumien kirjaamisessa, mikä käytännössä tarkoitti sitä, että jo olemassa olevia sairauksia tai koehenkilön sairaushistoriaan sisältyviä sairauksia ei kirjattaisi sen mukaan, miten päätutkija arvioi sairauden ”liittyvän luonteeseen”.
Tämä pöytäkirjassa oleva porsaanreikä, jonka täydellinen kuvaaminen on työläs työ, joka ei ole tämän mietinnön aiheena, on mahdollistanut huomattavan ”olosuhteiden huonontumisen” harhaanjohtamisen. Esimerkiksi koehenkilö 10791044 [135] näkee ”nivelrikon pahenevan”, minkä seurauksena hänen molemmat polvensa vaihdettiin, tai 11171146 aortan aneurysman poistuvan AE-lokista tutkimuslääkärin arvion perusteella. [136]
Todisteet siitä, että nämä kaksi tuotetta eivät ole täysin samanlaisia: markkinoille tulon jälkeisissä tutkimuksissa todettu kohonnut kuukautisvuotojen määrä
Myöhemmissä tutkimuksissa todettiin, että kuukautisvuotoa esiintyy paljon; esimerkiksi 12+% [137] Saudi-Arabiassa sijaitsevan yliopiston naisopiskelijoita ja henkilökuntaa koskevassa tutkimuksessa. Al Kadrin tekemässä meta-analyysissä [138] todettiin, että yhdistetty esiintyvyys oli 24,24 % (95 % CI: 12,8-35,6 %).
Tutkimuksen aikana [139] haittavaikutuksina oli raportoitu vain neljä kuukautisvuototapausta – kaksi BNT162b2-hoitoa saaneista (0,065 % 3087:stä 16-39-vuotiaasta naisesta).
Tarkasteltaessa sairaushistoriatietoja [140] löysimme 131 BNT:tä (1,77 %) 16-59-vuotiasta naishenkilöä, joilla oli aiemmin ollut menorragia – kaikkien kohdalla tapahtuman tilaksi oli asetettu ”VIIMEINEN TAPAHTUMA”. Osuus nousee 1,93 prosenttiin (143 tutkittavaa), jos mukaan otetaan muut tilat, joiden tutkimuslääkäri voi katsoa liittyvän menorragiaan (kohdun toimintahäiriöt, munasarjojen verenvuoto). Tämä on edelleen hyvin kaukana tutkimusten jälkeisistä luvuista, ja sen perusteella voidaan päätellä, että markkinoille saattamisen jälkeisissä tutkimuksissa todetut erittäin korkeat kuukautisvuototapausten luvut ovat toinen seuraus prosessiin 2 siirtymisestä.
Poikkeavuudet PCR-testauksessa
Tutkimuksessa oli yhteensä 110 päivää 27. heinäkuuta 2020 – ensimmäisestä satunnaistamisesta – 14. marraskuuta 2020 tapahtuneeseen tietojen katkaisuun, joka oli viimeinen VRBPAC:lle toimitettuun tietoon ja New England Journal of Medicine -lehdessä julkaistuun Polackin artikkeliin sisältyvä tietopiste. Tänä ajanjaksona, ensimmäisen annoksen jälkeen, kirjattiin 448 positiivista PCR-testiä plasebo-haarassa ja 163 positiivista testiä BNT162b2-haarassa [141].
Jos rajoitutaan protokollan mukaisesti testeihin, jotka on kirjattu 4 päivän kuluessa ilmenneillä oireilla, kokonaismäärä on 306 lumelääkettä ja 55 BNT162b2:ta. Oireettomuuden vuoksi hylätyn BNT162b2:n osuus (-66,3 %) verrattuna hylätyn lumelääkkeen määrään (-31,7 %) on luonnollisesti tilastollisesti erittäin merkittävä.
Korostimme 69 kohdassa COVID-oireiden uudelleenmäärittelyn vaikutusta. Sen vuoksi on hyödyllistä tehdä samanlainen ”oireanalyysi”, jossa otetaan huomioon kirjatut COVID-oireet riippumatta kliinisen tutkimuksen henkilökunnan ylläpitämästä tietokokonaisuudesta. Tämä näkökulma nostaa kokonaismäärät 70 oireiseen BNT162b2:een, joilla on positiivinen PCR (+15), 316:een Placeboa (+10) vastaan, eikä selitä epätasapainoa. Oireisten PCR-testien kertyminen kussakin haarassa on esitetty seuraavassa kaaviossa.
Analysoimme maittain [142] Yhdysvaltojen ja Argentiinan osalta niiden henkilöiden prosenttiosuuden, jotka ilmoittivat oireistaan ja joiden tuloksena tehtiin keskitetty PCR-testi 4 päivän kuluessa protokollan mukaisesti. Tasoitimme renderöinnin 3 päivän liukuvalla keskiarvolla. Seuraavasta kaaviosta käy ilmi, että Yhdysvalloissa kutakin ilmoitettua oireiden aaltoa seuraa merkittävä lasku molempien testauksessa. Vaikka on mahdollista, että testejä on ollut liian vähän suurten COVID-aaltojen aikana, on myös mahdollista, että testien manipulointia, kuten oireiden kirjaamisen lykkäämistä tai muita testien lieventämistapoja, on esiintynyt näiden jyrkkien testimäärän laskujen aikana, koska siitä ei ole riittävää dokumentaatiota. Tämä ei missään tapauksessa ole sellainen testausmalli, jota voisimme odottaa tutkimuksessa, jonka tavoitteena on tuottaa luotettavia tuloksia.
Sama havaittavissa oleva kuvio, eli testaustiheyden jyrkkä lasku voimakkaan oireiden raportoinnin jaksojen jälkeen, on nähtävissä – ja se tapahtuu myös Argentiinassa [143], kun raportoitujen oireiden määrä on vähäinen – mikä herättää lisäkysymyksiä tapahtumista, jotka vaikuttivat oireista raportoivien koehenkilöiden testaamiseen.
Koehenkilöiden COVID-käyntien PCR-testaukseen liittyvät poikkeavuudet
Vaiheen 3 satunnaistetut koehenkilöt tekivät yhteensä 10 219 ”oireenmukaista käyntiä” – kukin ”oireenmukainen käynti” vastaa oireiden perusteella epäiltyä COVID-tautia, mutta voi aiheuttaa useita fyysisiä käyntejä koehenkilön toimipaikassa useiden päivien välein [144].
Protokollaa noudattaen oireenmukaisten käyntien aikana otetut näytteet oli tarkoitus testata paikallisesti ja ”keskitetysti” Pfizerin Pearl Riverin laboratoriossa New Yorkissa, Yhdysvalloissa. Keskuslaboratoriotestit olivat suositeltavin menetelmä COVID-tapauksen vahvistamiseksi [145], vaikka protokollan vaihteluista riippuen voitiin käyttää FDA:n hyväksymiä paikallisia testejä [146]. Keskustestejä kirjattiin yhteensä 9474 ja paikallisia testejä 5489 kappaletta 13. maaliskuuta 2021 olevaan päättymispäivään mennessä.
BNT162b2-ryhmään kuuluvien henkilöiden 4421 oireenmukaisesta käynnistä 87,2 % johti PCR-testien lähettämiseen keskuslaboratorioon, kun taas plaseboryhmään kuuluvien henkilöiden 5798 käynnistä 88,3 %. Tämä pieni ero ei ollut merkittävä.
Sitä vastoin samana ajankohtana BNT162b2-ryhmän 4421 oireenmukaisesta käynnistä vain 43,5 % oli johtanut paikalliseen PCR-testaukseen, kun taas lumelääkeryhmän 5798 käynnistä 50,8 % oli johtanut paikalliseen PCR-testaukseen. Tämä ero on erittäin merkittävä, ja se osoittaa, että tietyissä paikoissa esiintyi merkittävä ero ryhmien välisessä hoidossa vastuullisten ryhmien toimesta.
On myös tärkeää tarkkailla PCR-testejä varten tehtyjen koehenkilöiden hoitoa oireenmukaisten käyntien yhteydessä kiireellisen käyttöluvan (EUA) päättyessä Yhdysvalloissa 14. marraskuuta 2020. Tähän päivämäärään mennessä oli kirjattu 5415 oireista käyntiä. Vain 65,1 % BNT162b2- ja 65,5 %:lla lumelääkepotilaista oireenmukaiset käynnit olivat johtaneet keskitettyyn testaukseen.
Vaikka ero paikallisten testien käsittelyssä on tilastollisesti vähemmän merkitsevä, se on edelleen olemassa. Tätä eroa, joka oli ilmeinen jo ennen kiireellisen myyntiluvan saamista, ei ole vielä selitetty.
Analysoitaessa oireenmukaisia käyntejä, jotka johtivat keskitettyihin PCR-testeihin protokollaikkunan sisällä [147], määräajassa tehtyjen testien määrä laskee 54,2 prosenttiin BNT162b2:n osalta ja 56,2 prosenttiin Placebon osalta. Määräajan kuluessa tehtyjen paikallisten testien osuus vähenee myös, mutta ryhmien välillä on edelleen merkittävä ero hoidon suhteen: BNT162b2:lla tehtiin 29,2 prosenttia testeistä ja Placebolla 33,3 prosenttia (p-arvo 8,103e-06).
Paikallisten testien analyysi koepaikoittain osoittaa, että sokeisiin kohdistuvat rikkomukset sijaitsevat Yhdysvalloissa
Palatakseni 127 kohdassa esitettyyn näkökulmaan (13. maaliskuuta 2021 COVID-käynnit – toisin sanoen BLA-tiedot analysoitiin), voimme keskittyä näihin poikkeavuuksiin testauspaikkakohtaisesti epänormaalien paikkojen tunnistamiseksi [148]. Suoritimme Fisherin tarkan testin koehenkilöille, joissa oli yhteensä vähintään 50 oireenmukaista käyntiä, jotta tunnistimme merkittävästi epätasapainoiset koehenkilöt — BNT162b2-ryhmän testaus oli systemaattisesti liian vähäistä verrattuna lumelääkeryhmään.
Arvioidaksemme, voisivatko harvinaisemmat, vaihtelevat tai vakavat oireet selittää kuilun, mittasimme t-testillä (t = -4,729, p-arvo = 2,451e-06) koehenkilöiden ilmoittamien oireiden ja käyntien välisen eron näillä koepaikoilla. Koehenkilöiden käynneillään ilmoittamien oireiden keskiarvo oli 2,49 BNT:llä ja 2,89 Placebolla, mikä osoittaa, että BNT-koehenkilöt ilmoittivat huomattavasti vähemmän oireita kuin Placebo.
Kun otetaan kuitenkin huomioon COVID-käynneistä johtuvien oireiden kyseenalainen luokittelu haittavaikutuksiksi, jota korostimme Augusto Roux’n todistajanlausunnon 46 kohdassa, ja ilmiön laaja-alaisuus, joka käy ilmi kohdista 61-66, on tarpeen huomata, että tämä vaikutus kääntyy päinvastaiseksi, jos tarkastelemme COVIDin kaltaisia oireita, jotka on raportoitu kaikissa tietokokonaisuuksissa. Seuraavassa kaaviossa esitetään niiden oireiden prosenttiosuus, jotka koehenkilöt ilmoittivat 7 päivän liukuvalla keskiarvolla molemmissa haaroissa ja jotka luokiteltiin COVID-oireiksi ja joihin liittyi COVID-käynti asianmukaisessa tietokokonaisuudessa. Se osoittaa, että BNT:n raportoimien oireiden osuus, jotka päättyivät oireenmukaiseen käyntiin, pysyi johdonmukaisesti alhaisempana aina EUA:n ja sokkoutuksen purkamiseen asti.
Yhteenveto tärkeimmistä poikkeamista tutkimuspaikkojen mittakaavassa
Merkittävimmät poikkeamat tutkimuspaikan mittakaavassa voidaan luokitella sokeisiin vaikuttaviin poikkeamiin:
havaintopoikkeamat, kohta 33
PCR-asteiden poikkeavuudet, kohta 131
ja ne, joissa on vakavia epäilyjä siitä, että henkilöitä on saatettu poistaa rekistereistä:
ilmoitettujen satunnaistamisnumeroiden poikkeavuudet, kohta 25
poikkeavuudet koehenkilöiden tunnistenumeroissa, kohta 48.
Seuraavissa taulukoissa esitetään yhteenveto kohteista, joihin on kohdistunut vaikutuksia; vain 17 kohdetta 153:sta luetellusta kohteesta on ”säästynyt” suurilta poikkeamilta.
Lähdeviitteet
1
FDA.gov – Ramachandra Naik, Ph.D et al. – Emergency Use Authorization (EUA) for an Unapproved Product Review Memorandum – https://www.fda.gov/media/144416/download
2
STATNews.com – Peter Doshi and Matthew Herder – Did the FDA understaff its review of the Pfizer/BioNTech vaccine? – https://www.statnews.com/2020/12/17/did-the-fda-understaff-its-review-of-the-pfizer-biontech-vaccine
3
PHMPT.org – UNITED STATES DISTRICT COURT FOR THE NORTHERN DISTRICT OF TEXAS FORT WORTH DIVISION – https://phmpt.org/wp-content/uploads/2022/01/ORDER_2022_01_06.pdf
4
PHMPT.org – Public Health and Medical Professionals for Transparency –
https://phmpt.org
5
R-4.4.0 for Windows – https://cran.r-project.org/bin/windows/base
6
R Script – download_full_prod.R – https://github.com/OpenVaet/pfizer_docs_R/blob/main/download_full_prod.R
7
R Script – extract_full_prod.R –
https://github.com/OpenVaet/pfizer_docs_R/blob/main/extract_full_prod.R
8
PHMPT.org – Pfizer 16+ Documents – https://phmpt.org/pfizer-16-plus-documents
9
pfizer.com – Pfizer and BioNTech to Submit Emergency Use Authorization Request Today to the U.S. FDA for COVID-19 Vaccine – https://www.pfizer.com/news/press-release/press-release-detail/pfizer-and-biontech-conclude-phase-3-study-covid-19-vaccine
10
FDA.gov – Susan Wollersheim, MD and Ann Schwartz, MD – BLA Clinical Review Memorandum – https://www.fda.gov/media/152256/download
11
R Script – extract_full_prod.R – screening_dates_range.R – https://github.com/OpenVaet/pfizer_docs_R/blob/main/screening_dates_range.R
12
PHMPT.org – Clinical Study Data Reviewer’s Guide – https://phmpt.org/wp-content/uploads/2022/05/125742_S1_M5_c4591001-S-csdrg.pdf, Page 24
13
PHMPT.org – 5.2-listing-of-clinical-sites-and-cvs-pages-1-41.pdf – https://phmpt.org/wp-content/uploads/2021/11/5.2-listing-of-clinical-sites-and-cvs-pages-1-41.pdf
14
R Script – screenings_by_weeks.R – https://github.com/OpenVaet/pfizer_docs_R/blob/main/screenings_by_weeks.R
15
Documentation – trial_site_data.json – https://github.com/OpenVaet/pfizer_docs_R/blob/main/trial_site_data.json
16
R Script – subjects_by_arms_and_sites.R – https://github.com/OpenVaet/pfizer_docs_R/blob/main/subjects_by_arms_and_sites.R
17
R Script – generate_screening_map.R, Scale: 1 subject / 100,000km² – https://github.com/OpenVaet/pfizer_docs_R/blob/main/generate_screening_map.R
18
PHMPT.org – C4591001 Investigational BNT162 Vaccine Program PF-07302048 – https://phmpt.org/wp-content/uploads/2022/03/125742_S1_M5_5351_c4591001-interim-mth6-oversight-committees.pdf, Page 42
19
FDA.gov – Susan Wollersheim, MD and Ann Schwartz, MD – BLA Clinical Review Memorandum – https://www.fda.gov/media/152256/download, Page 5
20
The New England Journal of Medicine – Edward E. Walsh, M.D et al. – Safety and Immunogenicity of Two RNA-Based Covid-19 Vaccine Candidates – https://www.nejm.org/doi/10.1056/NEJMoa2027906
21
Nature – Mark J. Mulligan et al., Phase I/II study of COVID-19 RNA vaccine BNT162b1 in adults – https://www.nature.com/articles/s41586-020-2639-4
22
R Script – screening_dates_range_phase1.R – https://github.com/OpenVaet/pfizer_docs_R/blob/main/screening_dates_range_phase1.R
23
PHMPT.org – Protocol C4591001 Protocol Amendment 9, 29 October 2020 – https://phmpt.org/wp-content/uploads/2022/03/125742_S1_M5_5351_c4591001-fa-interim-protocol.pdf, Page 87
24
PHMPT.org – Statistical Analysis Plan – https://phmpt.org/wp-content/uploads/2022/07/125742_S1_M5_5351_c4591001-fa-interim-sap.pdf, Pages 37 & 38
25
R Script – subjects_by_arms_and_sites_phase1.R – https://github.com/OpenVaet/pfizer_docs_R/blob/main/subjects_by_arms_and_sites_phase1.R
26
R Script – generate_screening_map_phase1.R, Scale: 1 subject / 100,000km² – https://github.com/OpenVaet/pfizer_docs_R/blob/main/generate_screening_map_phase1.R
27
PHMPT.org – 125742_S1_M5_5351_c4591001-interim-mth6-protocol.pdf – https://phmpt.org/wp-content/uploads/2022/03/125742_S1_M5_5351_c4591001-interim-mth6-protocol.pdf, Pages 1974, 1782, 1695, 32
28
PHMPT.org – 125742_S1_M5_5351_c4591001-fa-interim-iec-irb-consent-form.pdf, CONSENT TO TAKE PART IN STUDY – https://phmpt.org/wp-content/uploads/2022/04/125742_S1_M5_5351_c4591001-fa-interim-iec-irb-consent-form.pdf, Page 87
29
R Script – phase_1_adva.R – https://github.com/OpenVaet/pfizer_docs_R/blob/main/phase_1_adva.R
30
The New England Journal of Medicine – Edward E. Walsh, M.D et al. – https://www.nejm.org/doi/pdf/10.1056/NEJMoa2027906 – Page 10
31
Documentation – phase_1_different_adva_results.csv – https://github.com/OpenVaet/pfizer_docs_R/blob/main/phase_1_different_adva_results.csv
32
PHMPT.org – Analysis Data Reviewer Guide, 125742_S1_M5_c4591001-A-adrg.pdf – https://phmpt.org/wp-content/uploads/2022/03/125742_S1_M5_c4591001-A-adrg.pdf, Page 33
33
PHMPT.org – 2.7.1 Summary of Biopharmaceutic Studies and Associated Analytical Methods https://phmpt.org/wp-content/uploads/2022/06/125742_S1_M2_summary-biopharm.pdf, Page 3
34
PHMPT.org – Qualification of the SARS-CoV-2 mNeonGreen Virus Microneutralization Assay https://phmpt.org/wp-content/uploads/2023/04/125742_S1_M5_5314_vr-mqr-10214.pdf
35
PHMPT.org – Method Validation of the SARS-CoV-2 mNeonGreen Virus Microneutralization Assay https://phmpt.org/wp-content/uploads/2023/04/125742_S1_M5_5314_vr-mvr-10083.pdf
36
PHMPT.org – 125742_S1_M5_5351_c4591001-fa-interim-iec-irb-consent-form.pdf, CONSENT TO TAKE PART IN STUDY – https://phmpt.org/wp-content/uploads/2022/04/125742_S1_M5_5351_c4591001-fa-interim-iec-irb-consent-form.pdf, Page 67
37
PHMPT.org – 125742_S1_M5_5351_c4591001-fa-interim-investigators.pdf, 16.1.4.1 LIST OF INVESTIGATORS AND NUMBER OF SITES AND SUBJECTS BY THE COUNTRY – https://phmpt.org/wp-content/uploads/2023/10/125742_S1_M5_5351_c4591001-fa-interim-investigators.pdf
38
PHMPT.org – 125742_S1_M5_5351_c4591001-interim-mth6-investigators.pdf, 16.1.4.1 LIST OF INVESTIGATORS AND NUMBER OF SITES AND SUBJECTS BY THE COUNTRY – https://phmpt.org/wp-content/uploads/2023/10/125742_S1_M5_5351_c4591001-interim-mth6-investigators.pdf
39
R Script – investigators_screening_rando.R – https://github.com/OpenVaet/pfizer_docs_R/blob/main/investigators_screening_rando.R
40
Documentation – offset_randomization_between_fa_m6.csv – https://github.com/OpenVaet/pfizer_docs_R/blob/main/offset_randomization_between_fa_m6.csv
41
PHMPT.org – Analysis Data Reviewer Guide, 125742_S1_M5_c4591001-A-adrg.pdf – https://phmpt.org/wp-content/uploads/2022/03/125742_S1_M5_c4591001-A-adrg.pdf, Page 20
42
FDA.gov – Susan Wollersheim, MD and Ann Schwartz, MD – BLA Clinical Review Memorandum – https://www.fda.gov/media/152256/download, Page 7
43
The New England Journal of Medicine – Stephen J. Thomas, M.D. et al. – Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine through 6 Months – https://www.nejm.org/doi/10.1056/NEJMoa2110345 – “Between July 27, 2020, and October 29, 2020, a total of 45,441 participants 16 years of age or older underwent screening, and 44,165 underwent randomization at 152 sites”.
44
R Script – reproduce_rando_march_13.R – https://github.com/OpenVaet/pfizer_docs_R/blob/main/reproduce_rando_march_13.R
45
Documentation – phase_3_randomized_pop_stats.csv – https://github.com/OpenVaet/pfizer_docs_R/blob/main/phase_3_randomized_pop_stats.csv
46
PHMPT.org – Protocol C4591001 Protocol Amendment 9, 29 October 2020 – https://phmpt.org/wp-content/uploads/2022/03/125742_S1_M5_5351_c4591001-fa-interim-protocol.pdf, Page 57
47
R Script – format_deviations.R – https://github.com/OpenVaet/pfizer_docs_R/blob/main/format_deviations.R
48
PHMPT.org – Protocol C4591001 Protocol Amendment 9, 29 October 2020 – https://phmpt.org/wp-content/uploads/2022/03/125742_S1_M5_5351_c4591001-fa-interim-protocol.pdf, Page 64
49
FDA.gov – Ramachandra Naik, Ph.D et al. – Emergency Use Authorization (EUA) for an Unapproved Product Review Memorandum – https://www.fda.gov/media/144416/download, Page 14
50
PHMPT.org – Analysis Data Reviewer Guide, 125742_S1_M5_c4591001-A-adrg.pdf – https://phmpt.org/wp-content/uploads/2022/03/125742_S1_M5_c4591001-A-adrg.pdf, Pages 66 & 67
51
PHMPT.org – 125742_S1_M5_CRF_c4591001-1016-10161289.pdf – https://phmpt.org/wp-content/uploads/2023/05/125742_S1_M5_CRF_c4591001-1016-10161289.pdf, Page 270
52
Ema.europa.eu – ICH guideline for good clinical practice E6(R2) – Step 5 – https://www.ema.europa.eu/en/documents/scientific-guideline/ich-guideline-good-clinical-practice-e6r2-step-5_en.pdf, Section 4.11 – Safety Reporting, Section 4.9 – Records and Reports, Section 5.17 – Monitoring, Section 8 – Essential Documents for the Conduct of a Clinical Trial
53
PHMPT.org – 125742_S1_M5_CRF_c4591001-1231-12315632.pdf – https://phmpt.org/wp-content/uploads/2023/06/125742_S1_M5_CRF_c4591001-1231-12315632.pdf, Page 301 & 314
54
TheBMJ – Paul D Thacker – Covid-19: Researcher blows the whistle on data integrity issues in Pfizer’s vaccine trial – https://www.bmj.com/content/bmj/375/bmj.n2635.full.pdf
55
FDA.gov – https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/fda-bioresearch-monitoring-information/bioresearch-monitoring-program-information
56
davidhealy.org – Dr David Healy – Disappeared in Argentina – https://davidhealy.org/disappeared-in-argentina
57
IOS Press – David Healy, Augusto Germán Roux, Brianne Dressen – The coverage of medical injuries in company trial informed consent forms – https://content.iospress.com/articles/international-journal-of-risk-and-safety-in-medicine/jrs220043
58
Dr Hector E. Carvallo, Dr David Healy – Affidavit to Augusto Roux’s procedure
Doc1452332192
1.14MB ∙ PDF file
Download
59
R Script – subject_12312982_aes.R – https://github.com/OpenVaet/pfizer_docs_R/blob/main/subject_12312982_aes.R
60
R Script – verify_301.R – https://github.com/OpenVaet/pfizer_docs_R/blob/main/verify_301.R
61
PHMPT.org – Statistical Analysis Plan – https://phmpt.org/wp-content/uploads/2022/07/125742_S1_M5_5351_c4591001-fa-interim-sap.pdf, Pages 13 & 46
62
PHMPT.org – 125742_S1_M5_5351_c4591001-fa-interim-lab-measurements-sensitive.pdf, Table 16.2.8.2 – Listing of Subjects With First COVID-19 Occurrence From 7 Days After Dose 2 and Without Evidence of Infection Prior to 7 Days After Dose 2 – Evaluable Efficacy (7 Days) Population – https://phmpt.org/wp-content/uploads/2022/03/125742_S1_M5_5351_c4591001-fa-interim-lab-measurements-sensitive.pdf, Page 66 to 99
63
R Script – extract_official_EUA_cases.R – https://github.com/OpenVaet/pfizer_docs_R/blob/main/extract_official_EUA_cases.R
64
R Script – analyze_eua_official_cases.R – https://github.com/OpenVaet/pfizer_docs_R/blob/main/analyze_eua_official_cases.R
65
Youtube – Pfizer & The National Geographic – Mission Possible, Race for a Vaccine – https://youtu.be/jbZUZ9JYNBE, 00:30:13 to 00:31:17
66
Spectator, Australia – Jeyanthi Kunadhasan, MD – 170 patients that changed everything – https://www.spectator.com.au/2022/12/170-patients-that-changed-everything, archived at web.archive.org/web/20221208045723/https://www.spectator.com.au/2022/12/170-patients-that-changed-everything/
67
R Script – eua_cases_deviations.R – https://github.com/OpenVaet/pfizer_docs_R/blob/main/eua_cases_deviations.R
68
PHMPT.org – 125742_S1_M5_5351_c4591001-fa-interim-discontinued-patients.pdf, 16.2.1.4 Listing of Subjects Withdrawn From the Study – All Subjects – https://phmpt.org/wp-content/uploads/2022/06/125742_S1_M5_5351_c4591001-fa-interim-discontinued-patients.pdf, Page 78
69
Nature Communications – Antonio E. Muruato et al. – A high-throughput neutralizing antibody assay for COVID-19 diagnosis and vaccine evaluation – https://ncbi.nlm.nih.gov/pmc/articles/PMC7426916
70
MDPI – Ming Luo et al. – Nucleocapsid Structure of Negative Strand RNA Virus – https://www.mdpi.com/1999-4915/12/8/835
71
Oxford Academic – Michael D Swartz et al. – Antibody Duration After Infection From SARS-CoV-2 in the Texas Coronavirus Antibody Response Survey – https://academic.oup.com/jid/article/227/2/193/6581498
72
R Script – anayse_n_binding_pcr.R – https://github.com/OpenVaet/pfizer_docs_R/blob/main/anayse_n_binding_pcr.R
73
PHMPT.org – Annotated Study Book for Study Design: C4591001 – https://phmpt.org/wp-content/uploads/2022/05/125742_S1_M5_c4591001-S-acrf.pdf, Page 11
74
PHMPT.org – Clinical Study Data Reviewer’s Guide – https://phmpt.org/wp-content/uploads/2022/05/125742_S1_M5_c4591001-S-csdrg.pdf, Page 34
75
R Script – subjects_adverse_effects_deleted.R – https://github.com/OpenVaet/pfizer_docs_R/blob/main/subjects_adverse_effects_deleted.R
76
PHMPT.org – 125742_S1_M1_meeting-correspondence.pdf, 1.6.3 CORRESPONDENCE REGARDING MEETINGS – https://phmpt.org/wp-content/uploads/2023/10/125742_S1_M1_meeting-correspondence.pdf, Page 574
77
PHMPT.org – 125742_S1_M5_CRF_c4591001-1218-12181001.pdf – https://phmpt.org/wp-content/uploads/2023/05/125742_S1_M5_CRF_c4591001-1218-12181001.pdf, Page 401
78
PHMPT.org – 125742_S1_M5_CRF_c4591001-1087-10871286.pdf – https://phmpt.org/wp-content/uploads/2023/08/125742_S1_M5_CRF_c4591001-1087-10871286.pdf
79
R Script – aes_between_d2_d3.R – https://github.com/OpenVaet/pfizer_docs_R/blob/main/aes_between_d2_d3.R
80
PHMPT.org – CRFs for site 1055.pdf – https://phmpt.org/wp-content/uploads/2021/12/CRFs-for-site-1055.pdf, Pages 1125 to 1403
81
R Script – aes_crfs_missing.R – https://github.com/OpenVaet/pfizer_docs_R/blob/main/aes_crfs_missing.R
82
PHMPT.org – 125742_S1_M5_CRF_c4591001-1140-11401282.pdf – https://phmpt.org/wp-content/uploads/2023/08/125742_S1_M5_CRF_c4591001-1140-11401282.pdf
83
PHMPT.org – 125742_S1_M5_CRF_c4591001-1241-12411347.pdf – https://phmpt.org/wp-content/uploads/2023/08/125742_S1_M5_CRF_c4591001-1241-12411347.pdf
84
Koehenkilöt, joiden CRF-tiedostossa ei ole AE:tä, mutta jotka kuitenkin esiintyvät ADAE:ssä: 10081603, 10371141, 10371214, 10771137, 10811102, 10851018, 10901492, 10961181, 10961355, 11071196, 11101164, 11101165, 11171079, 11261244, 11281241, 11281267, 11331317, 11341327, 11351257, 11471230, 11501153, 11561131, 12314335, 12321299, 12411269, 12411493, 12411829, 12411930, 12412055, 12511029, 12511031, 12511033, 12511072, 12511145, 44441035
85
Koehenkilöt, joiden AE:t puuttuvat CRF-tiedostosta, mutta esiintyvät ADAE:ssä: 10071441, 10081152, 10131229, 10131386, 10131718, 10161087, 10191021, 10191145, 10211081, 10211084, 10281083, 10281205, 10381050, 10441163, 10471012, 10551128, 10551139, 10551145, 10551182, 10791183, 10801013, 10811179, 10831050, 10831173, 10841219, 10841538, 10871266, 10901140, 10911213, 10911247, 10911297, 10931067, 10931128, 11071191, 11091036, 11091074, 11091164, 11091204, 11101236, 11311140, 11331537, 11401066, 11401244, 11401282, 11401285, 11411143, 11461181, 11461302, 11521053, 11521095, 11521316, 11561001, 11781257, 11951017, 11951023, 12261477, 12312660, 12315186, 12315579, 12411053, 12411117, 12411347, 12411410, 12411561, 12411568, 12412191, 12412218, 12511060, 12541109, 12541189, 12601018, 12601108, 44441422
86
ECFR.gov – 21 CFR 312.57 – https://www.ecfr.gov/current/title-21/chapter-I/subchapter-D/part-312/subpart-D/section-312.57
87
Ema.europa.eu – ICH guideline for good clinical practice E6(R2) – Step 5 – https://www.ema.europa.eu/en/documents/scientific-guideline/ich-guideline-good-clinical-practice-e6r2-step-5_en.pdf
88
Documentation – adae_to_covid_symptoms.csv – https://github.com/OpenVaet/pfizer_docs_R/blob/main/adae_to_covid_symptoms.csv
89
PHMPT.org – 125742_S1_M5_5351_c4591001-interim-mth6-protocol.pdf – https://phmpt.org/wp-content/uploads/2022/03/125742_S1_M5_5351_c4591001-interim-mth6-protocol.pdf, Page 97
90
R Script – aes_and_symptoms_by_subjects.R – https://github.com/OpenVaet/pfizer_docs_R/blob/main/aes_and_symptoms_by_subjects.R
91
R Script – unblinding_by_arm_and_age_groups.R – https://github.com/OpenVaet/pfizer_docs_R/blob/main/unblinding_by_arm_and_age_groups.R
92
International Journal of Vaccine Theory, Practice, and Research – Corinne Michels et al. – Forensic analysis of the 38 subject deaths in the 6-Month Interim Report of the Pfizer/BioNTech BNT162b2 mRNA Vaccine Clinical Trial – https://ijvtpr.com/index.php/IJVTPR/article/view/86
93
Dr Jeyanthi Kunadhasan, MD (UKM), MMed (AnaesUM), FANZCA MMED (Monash), Open Letter to Dr Anthony Lawler, TGA’s Health Products Regulation Group – March 21, 2024
Amps Letter Tga March 21 2024
1.87MB ∙ PDF file
Download
94
Professor Anthony Lawler – TGA’s Health Products Regulation Group, Letter to Dr Jeyanthi Kunadhasan – March 27, 2024
D24 1170722 Response To Dr Jeyanthi Kunadhasan Australian Medical Professionals Society Re Undisclosed Deaths In C4591001trial 27 March 2024 1
103KB ∙ PDF file
Download
95
Dr Jeyanthi Kunadhasan, MD (UKM), MMed (AnaesUM), FANZCA MMED (Monash), Open Letter to Dr Anthony Lawler, TGA’s Health Products Regulation Group – May 17, 2024
Follow-up-letter-Professor-Anthony-Lawler_17_5_24.pdf
170KB ∙ PDF file
Download
96
The New England Journal of Medicine – Fernando P. Polack et al. – Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine – https://www.nejm.org/doi/full/10.1056/nejmoa2034577
97
Centers for Disease Control & Prevention – Underlying Cause of Death, 2018-2022, Single Race Request – https://wonder.cdc.gov/ucd-icd10-expanded.html, ”Year/Month: 2020; 2021”, ”Group By: State; Five-Year Age Groups; Year; Month”, ”Show Totals: Disabled”, ”Show Zero Values: False”, ”Show Suppressed: False”, ”Calculate Rates Per: 100,000”, ”Rate Options: Default intercensal populations for years 2001-2009 (except Infant Age Groups)”
98
Documentation – Underlying Cause of Death, 2018-2022, Single Race.txt – https://raw.githubusercontent.com/OpenVaet/pfizer_docs_R/main/us_mortality/Underlying%20Cause%20of%20Death%2C%202018-2022%2C%20Single%20Race.txt
99
Documentation – Single-Race Population Estimates 2020-2022.txt – https://github.com/OpenVaet/pfizer_docs_R/blob/main/us_mortality/Single-Race%20Population%20Estimates%202020-2022.txt
100
Centers for Disease Control & Prevention – Single-Race Population Estimates 2020-2022 Request – https://wonder.cdc.gov/single-race-v2022.html, ”Yearly July 1st Estimates: 2020; 2021”, ”Group By: State; Yearly July 1st Estimates; Age Group”, ”Show Totals: True”, ”Show Zero Values: False”
101
R Script – model_deaths_to_us_underlying_mortality.R – https://github.com/OpenVaet/pfizer_docs_R/blob/main/model_deaths_to_us_underlying_mortality.R
102
NEJM.org – Protocol – https://www.nejm.org/doi/suppl/10.1056/NEJMoa2034577/suppl_file/nejmoa2034577_protocol.pdf, Page 174
103
European Medicine Agency – Committee for Medicinal Products for Human Use (CHMP) – Product Assessment report (PAR) – https://www.ema.europa.eu/en/documents/assessment-report/comirnaty-epar-public-assessment-report_en.pdf, Page 18
104
Pharmaceutical and Medical Products Agency – Pharmaceutical Evaluation Division, Pharmaceutical Safety and Environmental Health Bureau, Ministry of Health, Labour and Welfare – Report on the Deliberation Results – https://www.pmda.go.jp/files/000243206.pdf, Page 13
105
PHMPT.org – Protocol C4591001 Protocol Amendment 9, 29 October 2020 – https://phmpt.org/wp-content/uploads/2022/03/125742_S1_M5_5351_c4591001-fa-interim-protocol.pdf, Page 310
106
PHMPT.org – Protocol C4591001 Protocol Amendment 9, 29 October 2020 – https://phmpt.org/wp-content/uploads/2022/03/125742_S1_M5_5351_c4591001-fa-interim-protocol.pdf, Page 360
107
PHMPT.org – Protocol C4591001 Protocol Amendment 9, 29 October 2020 – https://phmpt.org/wp-content/uploads/2022/03/125742_S1_M5_5351_c4591001-fa-interim-protocol.pdf, Page 360
108
Australian TGA, FOI 2389 – Pfizer/BioNTech COVID-19 mRNA vaccine (BNT162, PF-07302048) TGA Pre-Submission Meeting – https://www.tga.gov.au/sites/default/files/foi-2389-03-1.pdf, Pages 21 to 23
109
Australian TGA, FOI 3659 – BNT162b2 (PF-07302048) Comparability Report for PPQ Drug Product Lots – https://www.tga.gov.au/sites/default/files/2022-08/foi-3659-04.pdf, Pages 4 & 5
110
Australian TGA, FOI 3659 – BNT162b2 (PF-07302048) Comparability Report for PPQ Drug Product Lots – https://www.tga.gov.au/sites/default/files/2022-08/foi-3659-04.pdf, Page 3
111
PHMPT.org – 125742_S2_M3_32p5_batch-analyses.pdf, BNT162b2 3.2.P.5.4. Batch Analyses – https://phmpt.org/wp-content/uploads/2023/11/125742_S2_M3_32p5_batch-analyses.pdf, Page 3
112
PHMPT.org – 125742_S1_M5_5351_c4591001-interim-mth6-patient-batches.pdf – https://phmpt.org/wp-content/uploads/2022/06/125742_S1_M5_5351_c4591001-interim-mth6-patient-batches.pdf
113
Documentation – The Vaccine Adverse Event Reporting System (VAERS).txt – https://github.com/OpenVaet/pfizer_docs_R/blob/main/vaers/The%20Vaccine%20Adverse%20Event%20Reporting%20System%20(VAERS).txt
114
Centers for Disease Control & Prevention – VAERS – https://wonder.cdc.gov/vaers.html, VAERS Data Search, “Group by:Year Reported,Month Reported, Vaers Id”, “Vaccine Products: COVID19 (COVID19 VACCINE), COVID19-2 (COVID19-2)”, “Vaccine Manufacturer: Pfizer\BioNTech”, “Vaccine lot:EE8493, EJ0553”
115
Documentation – The Vaccine Adverse Event Reporting System (VAERS)_other_batches.txt – https://github.com/OpenVaet/pfizer_docs_R/blob/main/vaers/The%20Vaccine%20Adverse%20Event%20Reporting%20System%20(VAERS)_other_batches.txt
116
Medicines & Healthcare products Regulatory Agency, FOI 23/510 by Nick Hunt, Page 4
Ir 23 510
181KB ∙ PDF file
Download
117
PHMPT.org – 125742_S1_M5_5351_c4591001-fa-interim-patient-batches.pdf – https://phmpt.org/wp-content/uploads/2022/06/125742_S1_M5_5351_c4591001-fa-interim-patient-batches.pdf
118
R Script – process_2_subjects_by_sites_and_date.R – https://github.com/OpenVaet/pfizer_docs_R/blob/main/process_2_subjects_by_sites_and_date.R
119
Documentation – process_2_recipients_by_dates_and_sites.csv – https://github.com/OpenVaet/pfizer_docs_R/blob/main/process_2_recipients_by_dates_and_sites.csv
120
PHMPT.org – 125742_S1_M5_5351_c4591001-interim-mth6-randomization-sensitive.pdf, 16.1.7.1 Listing of Randomization Scheme and Actual Vaccine Received – All Subjects ≥16 Years of Age – https://phmpt.org/wp-content/uploads/2022/05/125742_S1_M5_5351_c4591001-interim-mth6-randomization-sensitive.pdf
121
R Script – process_2_subjects_by_randomization_numbers.R – https://github.com/OpenVaet/pfizer_docs_R/blob/main/process_2_subjects_by_randomization_numbers.R
122
R Script – compare_process_2_identification_methods.R – https://github.com/OpenVaet/pfizer_docs_R/blob/main/compare_process_2_identification_methods.R
123
R Script – model_process_2_aes.R – https://github.com/OpenVaet/pfizer_docs_R/blob/main/model_process_2_aes.R
124
MHRA’s Public Assessment Report for the Authorisation for Temporary Supply of Pfizer/BioNTech’s COVID-19 mRNA Vaccine BNT162b2 – https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/1112667/COVID-19_mRNA_Vaccine_BNT162b2__UKPAR___PFIZER_BIONTECH_ext_of_indication_11.6.2021.pdf
125
GOV.UK – ARCHIVE: Information for Healthcare Professionals on COVID-19 Vaccine Pfizer/BioNTech (Regulation 174) – https://www.gov.uk/government/publications/regulatory-approval-of-pfizer-biontech-vaccine-for-covid-19/information-for-healthcare-professionals-on-pfizerbiontech-covid-19-vaccine
126
Note that the PAR from December 2020 states a lymphadenopathy rate from doses 1 and 2 of 0.3% but the updated information for healthcare professionals cites a rate of 0.4%. The difference is because the original clinical trial had not finished recruiting participants when authorization was granted in December, 2020. The complete trial data put the rate at 0.4% instead of 0.3%.
127
European Journal of Nuclear Medicine and Molecular Imaging – Cohen, D., Hazut Krauthammer, S., Wolf, I. et al. – A sigh of relief: vaccine-associated hypermetabolic lymphadenopathy following the third COVID-19 vaccine dose is short in duration and uncommonly interferes with the interpretation of [18F]FDG PET-CT studies performed in oncologic patients. – https://doi.org/10.1007/s00259-021-05579-7
128
The New England Journal of Medicine – Moreira ED Jr, Kitchin N, Xu X, et al. – Safety and efficacy of a third dose of BNT162b2 Covid-19 vaccine. – https://www.nejm.org/doi/full/10.1056/NEJMoa2200674, Table 2
129
PHMPT.org – 125742_S1_M5_CRF_c4591001-1223-12231058.pdf – https://phmpt.org/wp-content/uploads/2023/05/125742_S1_M5_CRF_c4591001-1223-12231058.pdf
130
R Script – aes_cutoff_anomalies.R – https://github.com/OpenVaet/pfizer_docs_R/blob/main/aes_cutoff_anomalies.R
131
PHMPT.org – 125742_S1_M5_CRF_c4591001-1226-12261769.pdf – https://phmpt.org/wp-content/uploads/2023/06/125742_S1_M5_CRF_c4591001-1226-12261769.pdf
132
PHMPT.org – 125742_S1_M5_CRF_c4591001-1231-12312420.pdf – https://phmpt.org/wp-content/uploads/2023/06/125742_S1_M5_CRF_c4591001-1231-12312420.pdf
133
PHMPT.org – 125742_S1_M5_CRF_c4591001-1231-12313610.pdf – https://phmpt.org/wp-content/uploads/2023/06/125742_S1_M5_CRF_c4591001-1231-12313610.pdf
134
PHMPT.org – 125742_S1_M5_5351_c4591001-interim-mth6-protocol.pdf – https://phmpt.org/wp-content/uploads/2022/03/125742_S1_M5_5351_c4591001-interim-mth6-protocol.pdf, Page 206
135
PHMPT.org – 125742_S1_M5_CRF_c4591001-1079-10791044.pdf, https://phmpt.org/wp-content/uploads/2023/08/125742_S1_M5_CRF_c4591001-1079-10791044.pdf, Page 160
136
PHMPT.org – 125742_S1_M5_CRF_c4591001-1117-11171146.pdf, https://phmpt.org/wp-content/uploads/2023/05/125742_S1_M5_CRF_c4591001-1117-11171146.pdf, Page 178
137
R Script – inspect_menstrual_disorders_aes.R – https://github.com/OpenVaet/pfizer_docs_R/blob/main/inspect_menstrual_disorders_aes.R
138
MDPI, Medicina – Nahid Ibrahim Fallatah et al. – Menstrual Changes Following COVID-19 Vaccination: A Cross-Sectional Study – https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10890281/
139
Journal of Infection and Public Health – Hanan M. Al Kadri et al. – COVID-19 vaccination and menstrual disorders among women: Findings from a meta-analysis study – https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9979695
140
R Script – parse_medical_history.R – https://github.com/OpenVaet/pfizer_docs_R/blob/main/parse_medical_history.R
141
R Script – positive_pcrs_by_days_from_start.R – https://github.com/OpenVaet/pfizer_docs_R/blob/main/positive_pcrs_by_days_from_start.R
142
R Script – tests_on_symptoms.R – https://github.com/OpenVaet/pfizer_docs_R/blob/main/tests_on_symptoms.R
143
R Script – tests_on_symptoms_arg.R – https://github.com/OpenVaet/pfizer_docs_R/blob/main/tests_on_symptoms_arg.R
144
R Script – tests_on_symptomatic_visits.R – https://github.com/OpenVaet/pfizer_docs_R/blob/main/tests_on_symptomatic_visits.R
145
PHMPT.org – 125742_S1_M5_5351_c4591001-interim-mth6-protocol.pdf – https://phmpt.org/wp-content/uploads/2022/03/125742_S1_M5_5351_c4591001-interim-mth6-protocol.pdf, Page 63
146
PHMPT.org – 125742_S1_M5_5351_c4591001-interim-mth6-protocol.pdf – https://phmpt.org/wp-content/uploads/2022/03/125742_S1_M5_5351_c4591001-interim-mth6-protocol.pdf, Page 97
147
R Script – tests_on_symptomatic_visits_4_days.R – https://github.com/OpenVaet/pfizer_docs_R/blob/main/tests_on_symptomatic_visits_4_days.R
148
R Script – tests_on_symptomatic_visits_by_sites.R – https://github.com/OpenVaet/pfizer_docs_R/blob/main/tests_on_symptomatic_visits_by_sites.R
Artikkelin julkaissut openvaet.substack.com
http://eksopolitiikka.fi/ufot/pfizer-biontech-c4591001-koetutkimus-tarkastuskertomus-v1-2024-05-31/?utm_source=TR&utm_medium=eksopolitiikka.tumblr.com&utm_campaign=SNAP%2Bfrom%2B_%7C+Eksopolitiikka.fi+%7C_
0 notes
Text
Bill Gates ja WEF pyrkivät kriminalisoimaan rokotteesta kieltäytymisen
Maailman talousfoorumi (WEF) on yhdessä Bill Gatesin kanssa laatinut suunnitelman, jonka tarkoituksena on edistää pakollisia mRNA-rokotuksia sekä rajoittaa niiden kansalaisten oikeuksia, jotka kieltäytyvät rokotteista. Yksi keskeinen tavoite on estää Robert F. Kennedy Jr:n mahdollinen nimitys Yhdysvaltain terveysministeriksi, sillä hän on ollut väkevä ääni mRNA-rokotepakkoa vastaan.
Kennedy on ollut merkittävä kriitikko Bill Gatesin ja WEF:n ajamille rokotepolitiikoille, jotka sisältävät suunnitelmat pakollisista kuuden kuukauden välein annettavista mRNA-injektioista. Ei poikkeuksia, ei vapautuksia – vain totaalinen kontrolli. Gates ja WEF eivät pelkästään edistä näitä suunnitelmia, vaan myös pyrkivät kriminalisoimaan rokotteesta kieltäytymisen ja yhdistämään sen sosiaalisen luottopistejärjestelmän kanssa.
Rokotepassi vai syrjäytyminen?
Tulevaisuudessa rokotteesta kieltäytyminen saattaa tarkoittaa perusoikeuksien menettämistä. WEF:n visiossa kansalaiset, jotka eivät suostu ottamaan rokotteita, voivat menettää työpaikkansa, mahdollisuuden matkustaa tai käyttää peruspalveluita. Tämä malli on jo käytössä Kiinassa, jossa sähköautojen latausmahdollisuudet ja muut palvelut riippuvat kansalaisten sosiaalisesta luottopisteestä. Nyt tämä sama ajatus halutaan tuoda länsimaihin.
Tällainen kehitys ei ole vain uhka yksilön oikeuksille, vaan myös laajemmalle demokratialle. Hallitukset ja globaalit eliitit pyrkivät luomaan maailman, jossa erimielisyydet tukahdutetaan ja ainoa sallittu valuutta on tottelevaisuus.
Gatesin yhteydet ja tavoitteet
Bill Gates ei ole vain teknologia-alan miljardööri, vaan myös yksi suurimmista rahoittajista Maailman terveysjärjestössä (WHO). Gatesin vaikutusvalta ulottuu syvälle terveyssektoriin ja maailmanlaajuisiin rokoteohjelmiin. Hänen tavoitteensa näyttää olevan selkeä: saada kaikki ihmiset ottamaan mRNA-rokotuksia säännöllisesti ja varmistaa, ettei vastustajia jää jäljelle.
Gates on tavannut useaan otteeseen myös Donald Trumpin ja työskentelee aktiivisesti edistääkseen rokoteagendaa Yhdysvalloissa. Sisäpiiriläisten mukaan Gates on jopa rahoittanut operaatioita RFK Jr:n maineen tuhoamiseksi, ja hänen taktiikkansa muistuttavat monia aiemmin nähtyjä poliittisia likakampanjoita.
WHO:n pandemiasopimus ja tulevaisuuden uhkakuvat
WHO ja WEF haluavat pandemiasopimuksen, joka pakottaisi kaikki maat ottamaan käyttöön globaalin rokotepassijärjestelmän. Tämä tarkoittaisi, että suvereenit valtiot menettäisivät päätäntävaltansa terveystoimista ja siirtäisivät ne WHO:n ja sitä rahoittavien tahojen, kuten Gatesin, hallintaan.
Kennedy on yksi niistä harvoista poliitikoista, jotka ovat uskaltaneet vastustaa tätä kehitystä ja varoittaa kansalaisia tulevasta terveystyranniasta. Hänen sanomansa on selvä: ihmisten on puolustettava oikeuttaan päättää omasta kehostaan.
Heräämistä ilmassa
Huolimatta siitä, kuinka kovasti WEF ja Gates yrittävät työntää agendaansa läpi, ihmiset alkavat herätä tilanteeseen. Monet, jotka alun perin luottivat COVID-rokotteisiin, ovat alkaneet epäillä niiden tehokkuutta ja turvallisuutta. Rokotevastaisuutta ei enää voi leimata pelkäksi "salaliittoteoriaksi", sillä näemme jo nyt yrityksiä pakottaa kansalaiset ottamaan rokotukset vastoin tahtoaan.
Mikäli Gates ja WEF saavat tahtonsa läpi, tulevaisuus voi näyttää synkältä: digitaalinen identiteetti, sosiaaliset luottopisteet ja pakolliset rokotteet yhdistyvät yhdeksi suureksi valvontajärjestelmäksi.
Mutta ihmiset voivat vielä vastustaa. Nyt on aika kyseenalaistaa valtaapitelevien narratiivit ja vaatia oikeutta valita vapaasti ilman pelkoa syrjinnästä tai rangaistuksista.
Timo Tynkkynen
0 notes


Text
Kymen sanomat 14.10.2021


Mitä helvetin vittua? "Okei kato kun ongelma on se että me ei pystytä tekemään tarpeeksi rokotteita, nii ei siihen auta se, että annetaan muidenkin tehdä samanlaisia rokotteita" ????? Ööö, sehän siihen nimenomaan auttaa!?!?
Intiassa on tuotantokapasiteettia, joka ei pysty tällä hetkellä tekemään koronarokotteita, koska pfizerit ja modernat ovat tunkeneet rokotereseptinsä perseeseensä eivätkä suostu siihen et muut maat tekis heiän rokotteita.
Oikeesti, mikä helvetti rikkaita vaivaa? Onko olemassa mitään keinoa tehdä rikkaista oikeita ihmisiä? Auttaisko jos niiltä otettais rahat pois, tulisko niistä sitten taas oikeita ihmisiä, jotka ei olis niin... tollasia?
8 notes
·
View notes
Text
Rokotedenalialismi ei ollut isänmaallinen päätös.

Ilta-Sanomat 2021
2 notes
·
View notes
Photo
Neljän COVID-rokotteen Euroopassa julkisesti saatavilla olevan datan tarkastelua (30.3.2021)

Kirjoittanut: Catherine FRADE
Julkaistu: 1. huhtikuuta 2021

Lääketieteen tohtorina, entisenä lääketeollisuuden kansainvälisen sääntelyn johtajana haluan valaista ymmärrystänne virallisista tiedoista koskien neljää COVID-19 -rokotetta. Kaikki alla esitetyt tiedot on ladattavissa tästä linkistä. Olin innostunut työstäni laillisten tekstien kenttätutkimuksessa, jotta osaisin kehittää parhaiten lääkkeitä ja työskennellä synergiassa yrityksen ja terveysviranomaisten eri osastojen kanssa Ranskassa ja ulkomailla (jopa 83 maahan). 30. maaliskuuta 2021 Pfizer-, Moderna-, Astra Zeneca- ja Janssen-laboratoriot ovat saaneet ehdollisen myyntiluvan 4: lle COVID-19-rokotteelle (joulukuun 2020 ja maaliskuun 2021 välillä). Nykytilanteen ymmärtämiseksi on välttämätöntä käyttää lähdetietoja, kentällä toimimattoman henkilön on vaikea tunnistaa. Tämän artikkelin tarkoituksena on siis valaista julkista tietoa näistä neljästä COVID-19-rokotteesta viittaamalla kuhunkin lähteeseen, jotta niiden aitous voidaan vahvistaa. Tästä syystä löydät tästä artikkelista linkit myyntilupien virallisiin asiakirjoihin ja yhteenvedot Euroopan lääkeviraston (EMA) arvioimista tieteellisistä tutkimuksista .
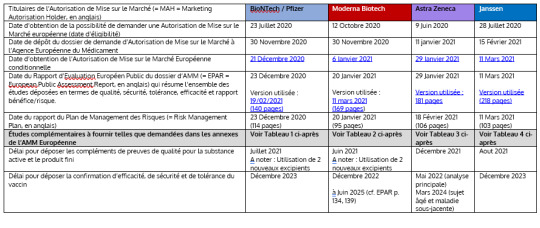
Ensinnäkin haluan selventää, että minulla ei ole eturistiriitoja lääketeollisuuden kanssa.
1. Ensinnäkin on tärkeää tietää, mikä ehdollinen myyntilupa on:
Myyntilupa (Marketing Authorization, MA/ML) myönnetään, kun tuote on osoittanut laadun, tehokkuuden ja turvallisuuden positiivisella hyöty-riskisuhteella (eli että sillä on enemmän hyötyjä kuin riskejä). Myyntiluvan saaminen on edellytys lääkkeen, mukaan lukien rokotteet, myymiselle. Kaikki MA-hakemuksen aikana tehdyt tutkimukset on tiivistetty EPAR-lausekkeeseen (= European Public Assessment Report). Se julkaistaan Euroopan lääkeviraston (EMA) verkkosivustolla. Mukana ovat myös suunnitellut tutkimukset, joita ei ole vielä tehty. Tämä aikataulu, joka kestää vuodet 2021-2025 COVID-19-rokotteesta riippuen, on määritelty ehdollisen myyntiluvan liitteissä ja EPAR-lausunnossa. Myyntilupa myönnetään myyntiluvan haltijalle (Marketing Authorization Holder, MAH). Ehdolliset myyntiluvat saatiin laadun, kliinisen ja ei-kliinisen tiedon perusteella rokotetestistä ja/tai bibliografisesta kirjallisuudesta. Bibliografisen kirjallisuuden tiedot ovat tunnustetuissa tieteellisissä lehdissä julkaistuja tutkimuksia, jotka on kirjoittanut myyntilupaa pyytäneen laboratorioon liittymättömät ja ulkopuoliset ryhmät. Nopeutetulla keskitetyllä menettelyllä saatu eurooppalainen myyntilupa sallii samanaikaisen markkinoinnin seuraavissa 30 maassa (Euroopan unioni ja Euroopan vapaakauppaliitto): Saksa, Itävalta, Belgia, Bulgaria, Kypros, Kroatia, Tanska, Espanja, Viro, Suomi, Ranska, Kreikka, Unkari, Irlanti, Islanti, Italia, Latvia, Liechtenstein, Liettua, Luxemburg, Malta, Norja, Alankomaat, Puola, Portugali, Romania, Slovakia, Slovenia, Ruotsi, Tšekki. Esimerkiksi Pfizerin EPAR (sivut 67 ja 114) päivämäärällä 19. helmikuuta 2021, joka on tähän mennessä eniten käytetty COVID-19-rokote, jolla on laajin käyttöindikaatio (yli 16-vuotiaat), mainitsee että tärkein ns. kliininen tutkimus on vaiheen 1/2/3 tutkimus, joka on edelleen kesken.
Alkuperäinen englanninkielinen EPAR Käännös Study C4951001: A Phase 1/2/3, Placebo-Controlled, Randomized, Observer-Blind, Dose-Finding Study to Evaluate the Safety, Tolerability, Immunogenicity, and Efficacy of SARS-COV-2 RNA Vaccine Candidates Against COVID-19 in Healthy Individuals The safety evaluation is based on one ongoing Phase 2/3 study that at the time of data cut-off (14-Nov-20) included 43,448 subjects who received either two doses of BNT162b2 30μg (n = 21,720) or placebo (n = 21,728). Overall, the reported reactogenicity profile are in line with any authorized vaccine. In addition, the frequency of reported AEs and SAEs were low. The emerging safety profile is presently considered favorable. Long term safety data, interaction with other vaccines, data on use in pregnancy and other subgroups (eg frail subjects, or subjects with pre-existing autoimmune diseases) are missing at this stage. The lack of long-term follow up renders the data provided non-comprehensive. Therefore, the delivery of the final C4951001 study report, including a 2-year follow up of the studied population, is classified as a specific obligation in the context of a conditional marketing authorization. The plan for the generation of further safety data post authorization is described in the section below. Tutkimus C4951001: Vaihe 1/2/3, lumekontrolloitu, satunnaistettu, tarkkailija-sokea, annosmääritystutkimus rokottajaehdokkaiden turvallisuuden, siedettävyyden, immunogeenisuuden ja tehokkuuden arvioimiseksi SARS-COV-2-RNA-rokotteelle COVID-19:a vastaan terveillä yksilöillä. (sivu 67) Turvallisuusarviointi perustuu käynnissä olevaan vaiheen 2/3 tutkimukseen, johon tietojen sulkemishetkellä (14.-20.11.) osallistui 43 448 potilasta, jotka saivat joko kaksi 30 μg: n annosta (n = 21 720) tai lumelääkettä (n = 21 728). Ilmoitettu reaktogeenisuusprofiili on kaiken kaikkiaan yhdenmukainen minkä tahansa lisensoidun rokotteen kanssa. Lisäksi ilmoitettujen haittavaikutusten ja vakavien haittavaikutusten esiintymistiheys oli pieni. Uusia turvallisuusprofiileja pidetään tällä hetkellä suotuisina. Pitkäaikaiset turvallisuustiedot, vuorovaikutus muiden rokotteiden kanssa, tiedot käytöstä raskauden aikana ja muut alaryhmät (esimerkiksi heikot tai koehenkilöt, joilla on jo autoimmuunisairauksia) puuttuvat tässä vaiheessa. Pitkän aikavälin seurannan puuttuminen tekee toimitetuista tiedoista tyhjentäviä. Siksi tutkimuksen C4951001 loppuraportin toimittaminen, mukaan lukien tutkimusväestön kahden vuoden seuranta, luokitellaan erityiseksi velvoitteeksi ehdollisen myyntiluvan yhteydessä. Suunnitelma ylimääräisten suojaustietojen tuottamiseksi valtuutuksen jälkeen on kuvattu alla olevassa osassa.
2. Yhteenvetotaulukko EMA:n verkkosivustolla julkaistusta neljän COVID-19-rokotteen myyntiluvasta:
Rekisteröintimenettelyn etenemisestä ja vielä toimitettavista tutkimuksista on oltava näkyvyys. Alla olevassa taulukossa on esitetty viralliset julkiset tiedot EMA-sivustolta: Nämä tiedot on otettu virallisista teksteistä 22. maaliskuuta 2021 (Alleviivatut tekstit viittaavat vastaaviin linkkeihin EMA:n virallisilla verkkosivuilla ja kaikki linkit on tiivistetty tämän asiakirjan loppuun).
Myyntiluvan haltijat (= MAH) BioNTech / Pfizer Moderna Biotech Astra Zeneca Janssen Päivämäärä, jolloin saatiin mahdollisuus hakea eurooppalaista myyntilupaa (kelpoisuuspäivä) 23. heinäkuuta 2020 12. lokakuuta 2020 9. kesäkuuta 2020 28. heinäkuuta 2020 Myyntilupahakemuksen jättöpäivä Euroopan lääkevirastolle 30. marraskuuta 2020 30. marraskuuta 2020 11. tammikuuta 2021 15. helmikuuta 2021 Ehdollisen eurooppalaisen myyntiluvan saamispäivä 21. joulukuuta 2020 6. tammikuuta 2021 29. tammikuuta 2021 11. maaliskuuta 2021 Päiväys, jona myyntiluvan haltijoiden asiakirja-aineistoa koskeva eurooppalainen julkinen arviointiraportti (= EPAR = englanniksi European Public Assessment Report) sisältää yhteenvedon kaikista toimitetuista tutkimuksista laadun, turvallisuuden, suvaitsevaisuuden, tehon ja hyöty-riskisuhteen suhteen. 23. joulukuuta 2020 Käytetty versio: 19.02.2021 (140 sivua) 20. tammikuuta 2021 Käytetty versio: 11. maaliskuuta 2021 (169 sivua ) 29. tammikuuta 2021 Käytetty versio: 181 sivua 11. maaliskuuta 2021 Käytetty versio (218 sivua) Riskienhallintasuunnitelman raportin päivämäärä 23. joulukuuta 2020 (114 sivua) 20. tammikuuta 2021 (95 sivua) 18. helmikuuta 2021 (106 sivua) 11. maaliskuuta 2021 (103 sivua) Lisätutkimukset toimitetaan Euroopan myyntiluvan liitteiden vaatimusten mukaisesti Katso alla oleva taulukko 1 Katso alla oleva taulukko 2 Katso alla oleva taulukko 3 Katso alla oleva taulukko 4 Määräaika tehoaineen ja lopputuotteen laatua koskevan lisätodistuksen toimittamiseksi Heinäkuu 2021 Huomaa: 2 uuden apuaineen käyttö Kesäkuu 2021 Huomaa: 2 uuden apuaineen käyttö Joulukuu 2021 Elokuu 2021 Määräaika rokotteen tehokkuuden, turvallisuuden ja sietokyvyn vahvistamisen toimittamiselle Joulukuu 2023 Joulukuu 2022 kesäkuuhun 2025 (vrt. EPAR s. 134, 139) Toukokuu 2022 (pääanalyysi) Maaliskuu 2024 (vanhukset ja perussairaus) Joulukuu 2023
3. Näiden julkisten tietojen analyysistä käy ilmi, että:
Nämä rokotteet ovat saaneet ehdollisen myyntiluvan, joka on voimassa yhden vuoden tavallisten myyntilupien viiden vuoden sijaan. Käynnissä olevat ja suunnitellut tutkimukset on saatava päätökseen, jotta voidaan hankkia vakiomuotoinen MA.
Vaikka kliiniset tutkimukset on suunniteltu alkavaksi, ne eivät ole täydellisiä, ja jotkut eivät ole vielä alkaneet. Rokotteesta riippuen lopulliset määräajat suunnitellaan vuosien 2022 ja 2025 välille (katso yllä oleva taulukko).
Yhden laboratorion COVID-19- rokotteen vaihdettavuudesta muiden laboratorioiden muiden COVID-19-rokotteiden kanssa rokotusaikataulun täyttämiseksi ei ole tietoa.
Käyttö rokote on tarkoitettu 18-vuotiaille , paitsi että Pfizerin ilmoitettu ikä on 16-vuotiaasta.
”Rokotteiden turvallisuutta ja tehoa lapsilla ja alle 18-vuotiailla nuorilla ei ole vielä varmistettu.” Tämä koskee Modernaa, Astra-Zenecaa ja Janssenia, joista ” tietoja ei ole saatavilla”. Sama Pfizerin tapauksessa ”lapsilla ja alle 16-vuotiailla nuorilla, joiden tietoja on rajoitetusti”.
Tietoa raskaana olevista naisista on hyvin rajallisesti (hylkäämiskriteerejä kliinisistä tutkimuksista): vähän tai ei lainkaan tietoa turvallisuudesta ja tehosta on toistaiseksi tiedossa. ( Taulukko 5 esimerkkinä). Raskaana olevien naisten rokottamista voidaan harkita vain tapauskohtaisesti. Pfizer-, Moderna- ja Janssen-rokotteiden osalta myyntiluvan liitteessä I esitetyssä tieteellisessä esitteessä (valmisteyhteenveto) ilmoitetaan, että Rokotteen käytöstä raskaana oleville naisille on vain vähän tietoja. Eläinkokeet eivät ole tuottaneet suoria tai epäsuoria haitallisia vaikutuksia raskauteen, alkion/sikiön kehitykseen, synnytykseen tai postnataaliseen kehitykseen (ks. AMM: n liitteessä I oleva 5.3 kohta). Käyttöä raskaana oleville naisille tulee harkita vain, jos mahdolliset hyödyt ovat suuremmat kuin mahdolliset riskit äidille ja sikiölle . ” Astra Zenecan pakkausselosteessa ei mainita vastaavia tietoja: ”Jos olet raskaana tai imetät, epäilet olevasi raskaana tai suunnittelet lapsen hankkimista, kysy neuvoa lääkäriltäsi, apteekista tai sairaanhoitajalta rokotteen ottamiseksi.”
Kokemuksen perusteella mikä tahansa markkinoille saatettu lääke antaa mahdollisuuden tuoda esiin laajamittaisia haittavaikutuksia, joita ei ole esiintynyt tai on harvoin esiintynyt kliinisissä tutkimuksissa. Tämä tarkoittaa konkreettisesti, että rokotuksiin liittyvät haittavaikutukset voivat ilmetä ajan myötä (mikä todennäköisesti selittää Astra-Zeneca-jakson maaliskuun puolivälissä 2021).
Nopeutetun keskitetyn eurooppalaisen menettelyn mukainen rokotteiden ehdollinen myyntilupa ”annettiin kansanterveyden eduksi täyttämään lääketieteellinen tarve”.
Jotkut professorit ja lääkärit ilmoittivat havainneensa alalla lääkeyhdistelmistä muodostettujen hoitojen tehokkuuden: viruslääkkeet, antibiootit, vitamiinit, ravintolisät jne. Tiedämme sen jälkeen tieteellisten julkaisujen erilaisesta kohtelusta käydyn keskustelun, joka voisi olla oikeudellisesti hyväksyttävää uusien rokotteiden ehdollisen myyntilupahakemuksen yhteydessä ja jota ei voida hyväksyä monien vuosien ajan käytettyjen lääkkeiden käytöstä (vrt. hydroksiklorokiini) …Jopa nykypäivänä asiasta edelleen debatoidaan.
Johtopäätös
Yhteenvetona voidaan todeta, että 30 maassa epätäydellisten ja/tai bibliografisten tutkimusten ja tulevien tutkimusten perusteella saatu ehdollinen eurooppalainen myyntilupa antaa ymmärtää, miten COVID-19-rokotteen antaminen vuonna 2021 on laajamittaisesti tutkittu. Ihmiset, jotka on rokotettu osana vielä kesken olevia tai tulevia tutkimuksia (kuten lapset, raskaana olevat naiset ja kaikki EPAR-taulukoissa esitetyt taulukot), joutuvat siksi kokeellisten tutkimus- ja kehitysprotokollien alaisiksi. Jokaisella on ennen rokotusta oikeus pyytää kaikki tietoisen suostumuksensa kannalta hyödylliset tiedot, mukaan lukien lääkityksen ohjeet (katso myyntiluvan liitteet, linkit tämän asiakirjan lopussa olevaan liitteeseen 6. Asiakirjat). Lisäksi ennalta varautumisen periaatteen on oltava vallitseva jo ennen mainitun suostumuksen saamista.

Tänään olen suuntautunut ammatillisessa toiminnassani ihmisten ja organisaatioiden terveyden ja elämänlaadun tukemiseen ja kouluttamiseen heidän tarkoituksensa mukaisesti, ja olen hyvin kiintynyt kaikkien kunnioittamiseen. Farmasian tohtorina ja kokonaisvaltaisena työpsykologina (CV liitteenä) yhdistän rationaaliset tieteet tietoisuuden tulokulmiin kaikissa muodoissaan. Näiden dokumentti- ja sääntelylähteiden tuella halusin antaa sinun löytää itsellesi hyvän ymmärryksen teksteistä ja siten valaista näitä uusia rokotteita. Dr. Catherine FRADE, ansioluettelo löytyy tästä. Farmasian tohtori, psykologi ja työpsykopatologi
Taulukko 1: Pfizer-myyntiluvan liite IIE, sivut 18 ja 19
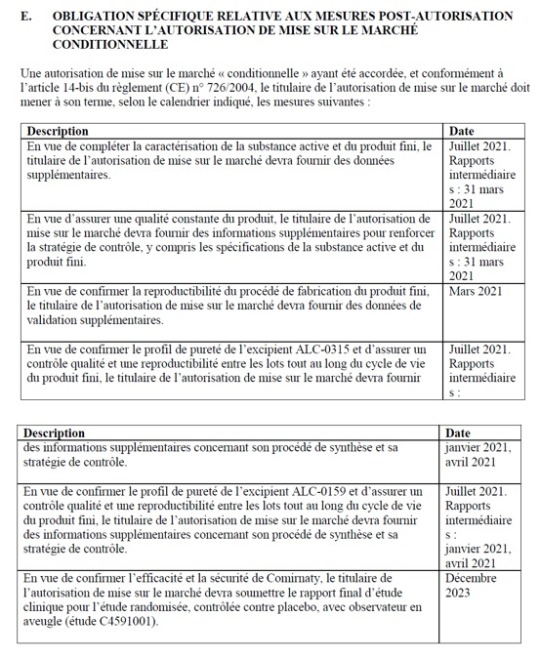
Taulukko 2: Modernan myyntiluvan liite IIE, sivu 15
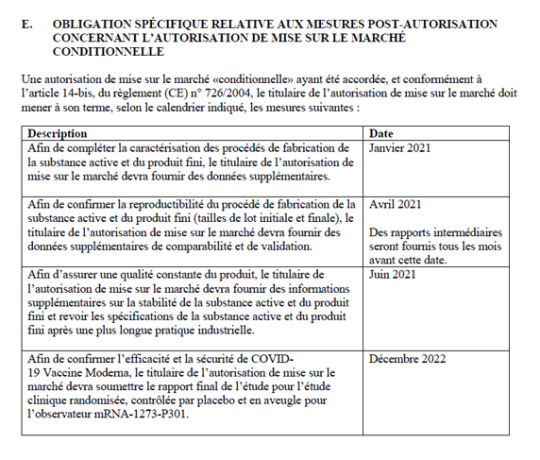
Taulukko 3: Astra Zenecan myyntiluvan liite IIE, sivut 14 ja 15
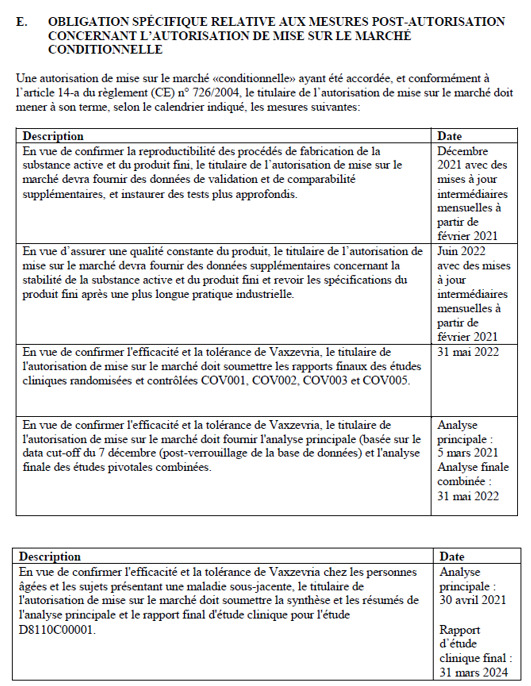
Taulukko 4: Janssenin myyntiluvan liite IIE, sivu 18
Taulukko 5: Raskaustiedot Pfizer EPAR:sta, esimerkkinä
Termi ”raskaana” tai ”raskaus” esiintyi EMA: n julkaisemassa EPAR-raportissa 10 sivulla . Raportin tarkat lauseet näkyvät sellaisina kuin ne ovat tässä taulukossa.
Extracts from the Pfizer EPAR – original text in English French translation by Deepl software Page 14. A study in pregnant women is also planned in the EU. A Post-Approval Active Surveillance Safety Study to Monitor Real-World Safety of Comirnaty (Study C4591010) will be conducted in the EU using primary data collection that monitors a cohort of vaccinees and evaluates risk of AESIs (adverse event of special interest) Sivu 14. Raskaana oleville naisille on suunnitteilla tutkimus myös EU: ssa. Hyväksynnän jälkeinen aktiivisen valvonnan turvallisuustutkimus Comirnatyn todellisen turvallisuuden seuraamiseksi (tutkimus C4591010) suoritetaan EU:ssa ensisijaisen tiedonkeruun avulla, joka seuraa rokotettujen kohorttia ja arvioi AESI-tautien (erityistä kiinnostusta aiheuttavan haittatapahtuman) riskiä. Page 68. 2.5.2. Main study, Title of study: Study C4951001: A Phase 1/2/3, Placebo-Controlled, Randomized, Observer-Blind, Dose-Finding Study to Evaluate the Safety, Tolerability, Immunogenicity, and Efficacy of SARS-COV-2 RNA Vaccine Candidates Against COVID-19 in Healthy Individuals Methods: Study Participants – exclusion criteria – Women who are pregnant or breastfeeding. Sivu 68. 2.5.2. Päätutkimus, tutkimuksen nimi: Tutkimus C4951001: Vaihe 1/2/3, lumelääkekontrolloitu, satunnaistettu, tarkkailija-sokea, annoshakututkimus SARS-COV-2-RNA-rokotteidenn turvallisuuden, siedettävyyden, immunogeenisuuden ja tehokkuuden arvioimiseksi terveillä henkilöillä COVID-19:a vastaan Menetelmät: Tutkimuksen osallistujat – poissulkemisperusteet – Raskaana olevat tai imettävät naiset. Page 93. Immunocompromised subjects and pregnant or breastfeeding women were excluded from the study. Sivu 93. Immuunipuutteiset potilaat ja raskaana olevat tai imettävät naiset suljettiin pois tutkimuksesta. Page 109. At the time of the data cut-off in the Phase 2/3 study (14 Nov 2020), a total of 23 participants had reported pregnancies in the safety database, including 9 participants who withdrew from the vaccination period of the study due to pregnancy. These participants are being followed for pregnancy outcomes. Thus, data on pregnancy are very limited at this stage. Sivu 109. Vaiheen 2/3 tutkimuksen tietojen katkaisuhetkellä (14. marraskuuta 2020) yhteensä 23 osallistujaa ilmoitti raskauksista turvallisuustietokannassa, mukaan lukien 9 osallistujaa, jotka vetäytyivät tutkimuksen rokotusjaksosta. raskauden vuoksi. Näitä osallistujia seurataan raskaustulosten suhteen. Siksi tiedot raskaudesta ovat tässä vaiheessa hyvin rajalliset. Page 113. 23 participants reported pregnancies in the safety database, nine of them were withdrawn from the study due to the pregnancy status. These participants will be followed up for pregnancy outcomes. Sivu 113. 23 osallistujaa ilmoitti raskauksista turvallisuustietokannassa, joista yhdeksän jätettiin pois tutkimuksesta raskaustilan vuoksi. Näitä osallistujia seurataan raskaustulosten suhteen. Page 114. clinical safety: Long term safety data, interaction with other vaccines, data on use in pregnancy and other subgroups (eg frail subjects, or subjects with pre-existing autoimmune diseases) are missing at this stage. Sivu 114. kliininen turvallisuus: Pitkäaikaiset turvallisuustiedot, yhteisvaikutus muiden rokotteiden kanssa, tiedot raskauden ja muiden alaryhmien käytöstä (esim. heikot tai koehenkilöt, joilla on jo autoimmuunisairauksia) puuttuvat tässä vaiheessa. Page 115. safety information: missing information = Use during pregnancy and while breast feeding Sivu 115. Turvallisuustiedot: puuttuvat tiedot = Käyttö raskauden ja imetyksen aikana Page 119. atypical COVID-19 in a cohort of people within the Department of Defense Healthcare System. For December 2023 Sivu 119. epätyypillinen COVID-19 puolustusministeriön terveydenhuoltojärjestelmän ihmisten joukossa. Joulukuussa 2023 Page 120. Study ( study short name, and title) Status (planned / on-going) : C4591015 Planned Summary of Objectives Planned clinical study to assess safety and immunogenicity in pregnant women who receive COVID-19 mRNA vaccine. Safety and immunogenicity of COVID-19 mRNA vaccine in pregnant women Safety concerns addressed: Use in pregnancy and while breast feeding. Protocol draft submission: 28-Feb-2021 Final CSR (Clinical study report) submission: 30-Apr-2023 Sivu 120. Tutkimus (tutkimuksen lyhyt nimi ja nimi) Status (suunniteltu / meneillään): C4591015 – tutkimus suunniteltu (ei vielä suoritettu) Yhteenveto tavoitteista: Suunniteltu kliininen tutkimus turvallisuuden ja immunogeenisuuden arvioimiseksi raskaana oleville naisille, jotka saavat COVID-19-mRNA-rokotetta. COVID-19-mRNA-rokotteen turvallisuus ja immunogeenisuus raskaana oleville naisille. Käsiteltyjä turvallisuuskysymyksiä: Käyttö raskauden ja imetyksen aikana. Protokollaluonnoksen toimittaminen: 28. helmikuuta-2021 Kliinisen tutkimusraportin lopullinen toimittaminen : 30. huhtikuuta-2023 Page 120. Study ( study short name, and title) Status (planned / on-going) : ACCESS / VAC4EU Planned Summary of Objectives Assessment of occurrence of safety events of interest, including severe or atypical COVID-19 in real-world use of COVID-19 mRNA vaccine. Safety concerns addressed: Anaphylaxis AESI-based safety events of interest including vaccine associated enhanced disease Use in pregnancy Use in immunocompromised patients Use in frail patients with co-morbidities (eg, chronic obstructive pulmonary disease (COPD), diabetes, chronic neurological disease, cardiovascular disorders) Use in patients with autoimmune or inflammatory disorders Protocol draft submission: 28-Feb-2021 Final CSR submission: 31-Jan-2024 Sivu 120. Tutkimus (tutkimuksen lyhyt nimi ja nimi) Status (suunniteltu / käynnissä): ACCESS / VAC4EU suunniteltu (ei vielä toteutettu) Yhteenveto tavoitteista Arvio kiinnostavien turvallisuustapahtumien esiintymisestä, mukaan lukien vaikea tai epätyypillinen COVID-19 COVID-19-mRNA-rokotteen tosiasiallisessa käytössä. Käsitellyt turvallisuuskysymykset: Anafylaksia Kiinnostavat turvallisuustapahtumat, jotka perustuvat erityisen kiinnostaviin haittatapahtumiin, mukaan lukien rokotteeseen liittyvät lisääntyneet sairaudet. Käytä raskauden aikana Käyttö immuunipuutteisilla potilailla Käyttö heikoilla potilailla, joilla on samanaikaisia sairauksia (esim. Krooninen obstruktiivinen keuhkosairaus (COPD), diabetes, krooninen neurologinen sairaus, sydän- ja verisuonisairaudet) Käyttö potilaille, joilla on autoimmuuni- tai tulehdussairaus. Pöytäkirjaluonnoksen toimittaminen : 28. helmikuuta 2021 Kliinisen tutkimusraportin lopullinen toimittaminen : 31. tammikuuta-2024 Page 136. Benefit-risk assessment and discussion 3.7.1. Importance of favorable and unfavorable effects There are no data on use in pregnant women, but a protective effect is anticipated. In the light of the reassuring data from the DART study, noting that pregnancy as such is a risk factor for severe COVID-19, and that pregnant women may additionally belong to other risk groups, vaccination may be considered on a case by case basis. Sivu 136. Hyöty-riskiarviointi ja keskustelu 3.7.1. Suotuisien ja epäedullisten vaikutusten merkitys Ei ole tietoa käytöstä raskaana oleville naisille, mutta suojaavan vaikutuksen odotetaan olevan. DART-tutkimuksen rauhoittavien tietojen valossa, jossa todetaan, että raskaus sinänsä on vakavan COVID-19:n riskitekijä ja että raskaana olevat naiset voivat lisäksi kuulua muihin riskiryhmiin, rokotuksia voidaan harkita tapauskohtaisesti.
Liite 6. Internetissä saatavilla olevat viitteet (ei tyhjentävä luettelo)
Aiheesta on olemassa suuri määrä erittäin mielenkiintoisia asiakirjoja:
Euroopan lääkevirasto (EMA): https://europa.eu/european-union/about-eu/agencies/ema_fr
Kansallinen lääkkeiden ja terveystuotteiden turvallisuusvirasto (ANSM): https://ansm.sante.fr/qui-sommes-nous/
Kysymykset ja vastaukset: Ehdollinen myyntilupa COVID-19-rokotteille EU:ssa: https://ec.europa.eu/commission/presscorner/detail/en/QANDA_20_2390
https://www.ema.europa.eu/en/news/meeting-highlights-pharmacovigilance-risk-assessment-committee-prac-8-11-march-2021
Asiakirjat Pfizer Moderna AstraZeneca Janssen Tiedot ovat saatavilla ilmaiseksi EMA: n kautta https://www.ema.europa.eu/en/medicines/human/EPAR/comirnaty https://www.ema.europa.eu/en/medicines/human/EPAR/covid-19-vaccine-moderna https://www.ema.europa.eu/en/medicines/human/EPAR/covid-19-vaccine-astrazeneca https://www.ema.europa.eu/en/medicines/human/EPAR/covid-19-vaccine-janssen Myyntiluvan liitteet https://www.ema.europa.eu/en/documents/product-information/comirnaty-epar-product-information_fr.pdf https://www.ema.europa.eu/fi/documents/product-information/covid-19-vaccine-moderna-epar-product-information_fr.pdf https://www.ema.europa.eu/fi/documents/product-information/vaxzevria-previously-covid-19-vaccine-astrazeneca-epar-product-information_fr.pdf https://www.ema.europa.eu/fi/documents/product-information/covid-19-vaccine-janssen-epar-product-information_fr.pdf EPAR-arviointikertomus – lääkärintarkastus https://www.ema.europa.eu/fi/documents/assessment-report/comirnaty-epar-public-assessment-report_en.pdf https://www.ema.europa.eu/fi/documents/assessment-report/covid-19-vaccine-moderna-epar-public-assessment-report_en.pdf https://www.ema.europa.eu/en/documents/overview/covid-19-vaccine-astrazeneca-epar-medicine-overview_fr.pdf https://www.ema.europa.eu/fi/documents/assessment-report/covid-19-vaccine-janssen-epar-public-assessment-report_en.pdf Yhteenveto lääkevalmistekomitean myönteisestä lausunnosta https://www.ema.europa.eu/en/documents/smop-initial/chmp-summary-positive-opinion-comirnaty_en.pdf https://www.ema.europa.eu/fi/documents/smop-initial/chmp-summary-positive-opinion-covid-19-vaccine-moderna_en.pdf https://www.ema.europa.eu/fi/documents/smop-initial/chmp-summary-positive-opinion-covid-19-vaccine-astrazeneca_en.pdf https://www.ema.europa.eu/fi/documents/smop-initial/chmp-summary-positive-opinion-covid-19-vaccine-janssen_en.pdf Riskienhallintasuunnitelma https://www.ema.europa.eu/en/documents/rmp-summary/comirnaty-epar-risk-management-plan_en.pdf https://www.ema.europa.eu/fi/documents/rmp-summary/covid-19-vaccine-moderna-epar-risk-management-plan_en.pdf https://www.ema.europa.eu/fi/documents/rmp-summary/covid-19-vaccine-astrazeneca-epar-risk-management-plan_en.pdf https://www.ema.europa.eu/documents/rmp-summary/covid-19-vaccine-janssen-epar-risk-management-plan_en.pdf
Tämä artikkeli lähetettiin myös:
https://www.linkedin.com/posts/catherinefrade_%C3%A9clairage-sur-les-donn%C3%A9es-publiques-europ%C3%A9ennes-activity-6783385465841082368-KqWr Blogissani täällä: http://www.catherinefrade.com/blog/2021/04/01/ecclairage-sur-les-donnees-publiques-europeennes-des-amm-conditionnelles-pour-les-4-vaccins-covid -19-31-maaliskuu-2021 / Artiklan 2 mukaan Choletin sairaalan CTIAP on 2. huhtikuuta 2021: http://ctiapchcholet.blogspot.com/2021/04/inedit-exclusif-vaccins-contre-la-covid.html 3 artikla, joka on yhdenmukainen oikeusvaikutuksia koskevan 1 artiklan kanssa: https://www.linkedin.com/posts/catherinefrade_vaccins-covid19-vaccination-activity-6785286370953895936-TqHn Eri verkkosivustojen välittämä 1 artikla: https://www.medias-presse.info/doute-sur-la-qualite-intrinseque-des-produits-presentes-comme-vaccins-anti-covid-et-leurs-procedes-de-fabrication-sur-base- virallisten asiakirjojen julkaisema-Euroopan-lääkeviraston / 141451 / saksaksi: Zweifel an der intrinsischen Qualität von Produkten, die als Anti-Covid-Impfstoffe präsentiert werden | MITTELEUROPAN ALALLA https://www.linkedin.com/posts/chantalattia_ces-vaccins-sont-une-exp%C3%A9rimentation-d%C3%A9lai-activity-6784593281922695168-u10g
Jos tiedät muita artikkelin loppuosia, ilmoita minulle, että voin myös jakaa. Ota yhteyttä saadaksesi lisätietoja virallisten julkisten tietojen tulkinnasta.
Lähde:
https://kapitaali.com/neljan-covid-rokotteen-euroopassa-julkisesti-saatavilla-olevan-datan-tarkastelua-30-3-2021/
0 notes
Text

62 notes
·
View notes
Text
Homeet ja rokotteet - paha yhdistelmä
Homeet ja rokotteet – paha yhdistelmä
Kirjoittaja: Christer Sundqvist
Suomen ME/CFS yhdistys (SMEY) on ottanut kantaahyvin ajankohtaiseen terveysongelmaan. Sain kuulla, ettei oikein kukaan viitsi tai uskalla tästä aiheesta kirjoittaa. Turpaduunari uskaltaa! Hän on pelkkä yksinkertainen biologi, joka harjoittaa ammattiaan niin hyvin kuin se on mahdollista. Lokaa tulee niskaan, mutta terveyden ammattilaisista poiketen Valvirat, AVI sun…
View On WordPress
#Christer Sundqvist#home#kipu#krooninen väsymysoireyhtymä#ME/CFS#monikemikaaliherkkyys#rokotteet#sisäilmaongelmat#turpaduunari#tutkimus
0 notes
Text
Innovointi blogi
Innovaatio tarkoittaa uutta tai olennaisesti paranneltua taloudellelisesti hyödyllistä tuotetta. Innovointi voi olla jonkinlainen esine, idea tai käytäntö. Innovaatio voi olla myös nopeasti asian mullistava keksintö tai vähän kerrallaan, mutta pitkällä aika välillä tehty muutos tai parannus.
Tavallinen ihminen huomaa yleensä erilaisia innovaatioita siinä kun isot yhtiöt puskevat markkinoilla paljon erilaisia ja paranneltuja tuotteita esimerkiksi erilaisia paranneltuja älykännyköitä tulee markkinoille vuosittain.
Innovointi on tärkeää, koska ilman sitä kehitys lakkaa ja pysyisimme samalla tasolla esimerkiksi teknologiassamme. Monet yritykset myös kaatuisivat ilman innovointia, sillä jos uusia tuotteita ei tule markkinoille loppuu, myös ihmisten mielenkiinto kyseistä yhtiötä kohtaan.
Innovaatiot kuten esimerkiksi erilaiset rokotteet ja lääkkeet ovat myös pelastaneet paljon ihmisiä ja muutenkin ihmisten elinajanodote kasvaa kokoajan tämän vuoksi
1 note
·
View note
Text
Koronasta ja koronapassista
– Käyttäkää maskia niin päästään koronasta.
Ei päästy.
– Ottakaa rokote niin päästään koronasta.
Ei päästy.
– Käyttäkää taas maskia ja ottakaa toinen rokote niin päästään koronasta.
Ei päästy.
– No, käyttäkää koronapassia niin päästään koronasta.
Ei päästy.
– No hei, ottakaa kolmas rokote...
Kestin koronaa ihan hyvin viime vuoden ja vähän kipuillen tämän vuoden, mutta näyttää siltä että tämä ei nyt ota vaan loppuakseen. Ainoa ratkaisu mitä meille tarjotaan on koronapassi ja jatkuva sarja rokotteita tai niiden tehosteita, vaikka kumpikaan näistä ei näytä laskeneen tartuntamääriä. Määrät on paremminkin nousussa.
Kun omikronista uutisoitiin Etelä-Afrikassa, tuli se Pohjoismaihin muutamassa päivässä ja sai kaikki tartuntaennusteet räjähtämään. Nyt Pohjoismaissa on peloteltu, että ensi vuonna tartuntoja voi tulla jopa satoja tuhansia päivässä.
Mun mielestä ongelma tällä hetkellä on se että päättäjät ja ihmiset luottavat liikaa koronapassiin. Koronapassi antaa ihmisten ymmärtää että nämä voivat turvallisesti kokoontua missä vain, vaikka tiedetään että rokotteet eivät edes estä saamasta tartuntaa tai tartuttamasta muita. Rokote suojaa vain sua itseäsi taudin vakavalta muodolta, mutta ei tee juuri mitään muuta.
Se mitä olisi pitänyt tehdä jo vuoden ja pitäisi tehdä edelleen on testaaminen. Joka ikiseltä Suomeen tai mihin vain vieraaseen maahan saapuvalta pitäisi vaatia negatiivinen testitulos riippumatta rokotteiden määrästä. Samoin jokaiselta yleisiin tilaisuuksiin ja tapahtumiin menevältä pitäisi vaatia negatiivinen testitulos. Tuo on ainoa asia, jolla voidaan olla 99% varmoja että henkilö ei tartuta tautia eteenpäin vaikka olisi altistunut.
Koronapassi pitäisi mun mielestä myöntää pelkästään negatiivisella tuloksella väliaikaisesti. Muuten tästä riesasta ei päästä vuosiin. Miksi Suomen päättäjät tai terveydenhuollon viranomaiset eivät tätä halua ymmärtää? En halua kuulostaa miltään foliohatulta, mutta tulee sellainen tunne että tässä halutaan vain myydä lyhytkestoisia rokotteita, jotka suojaavat vakavalta taudilta mutta eivät estä viruksen leviämistä ja pandemian pitkittymistä. Lääkefirmojen etu on, mitä pidempään pandemia saadaan pidettyä päällä.
Musta kuitenkin tuntuu että tässä mennään ihan eri suuntaan. Ens vuonna varmaan vaaditaan koronapassiin kolme rokotusta ja sitä ei enää saa testeillä tai sairastetulla koronalla ollenkaan. Sanokaa mun sanoneen. Sitten ihmetellään taas kun tartuntamäärät vaan kasvavat ja pitää vetää “hätäjarrusta” joka toinen kuukausi ja laittaa klubit ja ravintolat konkurssiin.
Mä haluan matkustaa ja päästä keikoille ja tapahtumiin. Haluan viettää normaalin nuoruuden enkä asua tynnyrissä parhaita vuosiani. Alkaa ihan oikeasti riittää. Ennusteet siitä että tää paska kestäis vielä 4-5 vuotta saa mut voimaan fyysisesti pahoin.
1 note
·
View note
Text
Rokotteet ja geenimutaatio
Rokotteet ja geenimutaatio

kirjoittanut Harold E. Buttram, MD; Susan Kreider, RN; Alan R. Yurko lokakuu 11, 2002
Johdanto
Tämä artikkelin kirjoittajat eivät esitä olevansa auktoriteetteja genetiikan tai immunologian aloilla. Maallikkona olossa on se etu, että kun asiaa tarkastelee etäältä, voi joskus havaita asioita jotka jäävät asiantuntijoilta huomaamatta, jotka ovat kiinni oman alansa yksityiskohdissa ja monimutkaisuuksissa. Tämä voi olla totta puhuttaessa rokotteiden vaikutuksista immunologiaan ja genetiikkaan, joista tiede oikeasti tietää aika vähän.
Tämä artikkeli käy läpi kolmen alan asiantuntijan, LT John Martinin, tri. Howard B Urnovitzin ja tri. MG Montinarin työtä, joista käy ilmi varsin vakuuttavat näytöt sille, että joiltain rokotteiden saaneilta potilailta löytyy geneettisiä muutoksia, joiden reaktiot vaikuttavat liittyvän rokotteiden kausaalimekanismiin. Tässä ei esitetä väitteitä, että tämä evidenssi olisi vedenpitävä todiste rokotteiden aiheuttamista geneettisistä muutoksista. Se mitä me toivomme esittävämme on näiden tutkijoiden työ, eli että on sekä mahdollista että uskottavaa, että nykyiset lapsuusajan rokoteohjelmat saattavat aiheuttaa laajamittaisia geenimuutoksia, mikä mahdollisesti vaikuttaa laajalti omiin lapsiimme.
Todistustaakka rokotereaktioista ei tulisi levätä vanhempien harteilla, niinkuin se tällä hetkellä lääketieteellis-juridisessa systeemissämme on. Todistustaakka rokotteiden turvallisuudesta; eli että rokotteet EIVÄT aiheuta negatiivisia geenimuutoksia, tulisi olla valmistajien, liittovaltion ja valtion viranomaisten sekä tällä hetkellä rokotteita antavien koulujen harteilla. Ennen kuin tämä asia on selvitetty, onko kenelläkään millään tasolla yhteiskuntaamme oikeutta pakottaa yhä vain suurempaa määrää rokotteita lapsillemme?
Perustietoa immunologiasta
Vaikka ihmisen immuunijärjestelmän tekniset yksityiskohdat ovat äärimmäisen monimutkaisia, perusoperaatiot ovat olennaisesti yksinkertaisia ja niitä voitaisiin verrata keskiaikaiseen linnaan. Tätä analogiaa käyttäen, ensin saattaa olla muutama etuvartioasema, sitten vallihauta, sitten päärakennuksen seinä, ja lopulta sisemmät puolustukset linnan ympärillä, jossa sisällä kuninkaalliset asuvat. Kuninkaalliset tässä tarkoittavat ihmisen geneettistä järjestelmää, jota immuunijärjestelmä on suunniteltu suojelemaan kaikin tavoin.
Etuvartiot tarkoittaisivat lymfosyyttejä (yhdenlaisia valkosoluja), joita kutsutaan “muistisoluiksi” koska ne muistavat tunkeutujille altistumisen. Altistuminen käynnistää kloonausprosessin tulevien altistusten varalta. Päärakennuksen seinämää esittää hengitysteiden ja suolen limakalvot, ja sisäpuolustusta vasta-aineita tuottavat veren plasmasolut (ja muut valkosolut), jotka ovat luuytimessä.
Immuniteetti
Soluvälitteinen immuniteetti
Ihmisevoluution lukemattomien vuosituhansien aikana limakalvojen soluvälitteinen immuniteetti ihmiskehossa on ollut pääasiallinen sisääntuloreitti sairauksia aiheuttaville mikro-organismeille ihmiskehossa, ja täten evoluution myötä limakalvot ovat kehittäneet kehoon suuren puolustusjärjestelmän. Nämä limakalvot on pinnoitettu “antiseptisella pinnoitteella”, joka koostuu miljardeista ja biljoonista molekyyleista immunoglobuliini A -vasta-aineita, joiden rooli on tunnistaa jokainen ohitse kulkeva molekyyli, sisäelinten tapauksessa jaotella ravintoaineet, ja pysäyttää kaikki tunnistamattomat aineet, mm. epätäydellisesti sulaneet ruoka-aineet. Vaadittaisiin useita suuria tietokoneita pääsemään tämän järjestelmän älykkyyden tasolle. Kaikki toimii erittäin ohuen (ja tyhmän) vatsan limakalvon sekä useiden myrkyllisten aineiden välillä, jotka muuten menisivät läpi limakalvosta verenkiertoon. Antiseptisen pinnoitteen lisäksi limakalvojen puolustusmekanismin päätoimija infektioita aiheuttavia mikro-organismeja vastaan on soluvälitteinen immuniteetti, jonka vaikuttavat aineet ovat fagosyyttiset makrofaagit (syöjäsolu) ja sytotoksiset T-lymfosyytit (imusolu).
Vasta-aineimmuniteetti
Sisäisiä puolustusmekanismeja esittää plasmasolut luuytimessä, jotka tuottavat vasta-aineita, mikä normaalisti toimii sekundäärisenä kehon puolustusmekanismina. Se käynnistyy limakalvojen soluimmuniteetin lisänä, tai pääasiallisena puolustusmekanismina kun soluimmuniteetti on pettänyt. Tähän immuniteettiin viitataan vasta-aineimmuniteettina. Plasmasolut voivat tuottaa
(1) Makroglobuliineja, jotka ilmaantuvat akuutin infektion esiintyessä ensimmäisenä, jotka ovat primitiivisempiä ja jotka toimivat yleisluonnollisena antibioottina;
(2) Immunoglobuliini G vasta-aineet, jotka ovet erittäin spesifisiä tiettyä ulkopuolista tunkeutujaa vastaan ja ilmaantuvat jonkin verran myöhemmin infektion edetessä kloonausprosessin käynnistyttyä ja
(3) IgE vasta-aineet, jotka saavat aikaan allergioita.
Lastentautien rooli
On olemassa koulukunta, jonka mielestä niinkutsutut entisaikojen lastentaudit, mm. erilaiset rokot (tuhka-, vesi- jne.), jotka ovat päässeet kehoon limakalvojen kautta, ovat olleet tarpeellisia ja niillä on positiivinen tarkoitus näiden limakalvojen immuunijärjestelmän kehittämisessä ja vahvistamisessa. Rokotteet sitä vastoin injektoidaan suoraan kehoon, mikä ohittaa limakalvot ja jättää näiden kautta syntyvän immuniteetin suhteellisen heikoksi ja kitukasvuiseksi.
Sekä The New England Journalissa (1) että Thorax-julkaisussa (2) on julkaistu artikkeleja, joissa sanotaan että terveellä immuunijärjestelmällä on “bias” soluvälitteistä immuniteettia kohtaan, kun taas ihmisillä joilla on allergioita, astmaa, autoimmuunisairauksia on vasta-ainedominantti immuunijärjestelmä. On myös osoitettu, että kun jompi kumpi näistä tulee dominantiksi, on vaikeaa vaihtaa järjestelmästä toiseen. (3)
Geenien vaihdanta ympäristön kanssa
Barbara McClintock, vuonna 1983 Nobel-palkittu “Corn Lady”, ensimmäisenä havaitsi niinkutsuttujen hyppygeenien geneettisen liikkuvuuden 1930-luvulla. Yli 50 vuotta hän teki yksin tutkimusta maissin parissa, ja selvitti erään luonnon kaikkein syvimmistä salaisuuksista.
McClintock tutki intiaanimaissia, jossa punaisten ja keltaisten maissinjyvien jakauma määräytyy geneettisesti. Hän havaitsi, että jotkut geenit liikkuivat paikasta toiseen solun kromosomeissa (kuin rukousnauhan helmet). Sitten hän näki liikekuvioita, jotka erosivat kovin värillisten jyvien liikkeistä, ja tajusi että jotkut geenit, kun ne ovat kulkeutuneet tiettyyn paikkaan, kytkivät geenejä päälle tai pois. Siitä seuraa, että vaikka useimmat geenit ovat työläisiä, jotkut geenit olivat näiden johtajia.
World Medicinen artikkelin mukaan (syyskuu 22, 1971, s. 69-72; New Medical Journals, Clareville House, Oxendon St., London), Genevan yliopiston tutkijat olivat tehneet hämmästyttävän havainnon, että verenkiertoon päätyvät biologiset aineet muuttuvat todella osaksi meitä ja jopa geenejämme. Artikkelissa sanotaan:
“Kun japanilaiset bakteriologit saivat selville, että yhden lajin bakteerit siirsivät oman erittäin omalaatuisen antibioottivastustuskykynsä kokonaan täysin toiseen lajiin, he vaikuttivat törmänneensä johonkin täysin uniikkiin ellei sitten hämmästyttävään ilmiöön. Tri. Maurice Stroun ja tri. Pilippe Anker, yhdessä kollegoidensa kanssa kasvifysiologian laitokselta Genevan yliopistosta keräsivät mittavasti todisteita siitä, että geneettisen informaation siirtäminen ei rajoitu pelkästään bakteereihin, vaan se voi tapahtua myös bakteerien välillä sekä kasveilla ja eläimillä.”“Genevalaiset tieteentekijät ovat vakuuttuneita, että normaali eläin- ja kasvisolu voi myös levittää DNA:ta ja että tämä DNA kulkeutuu muihin soluihin organismissa. Jos he ovat oikeassa, silloin sillä on merkittävästi vaikutusta käytännössä jokaiseen solun aineenvaihdunnan aspektiin. Se vaikuttaisi organismin kasvuun ja kehitykseen, sairauksiin ja jopa organismin evoluutioon.” “Tri. Maurice Stroun ja hänen kollegat tekivät suurimman osan tutkimuksista kasveilla, mutta he ovat nyt siirtyneet eläimiin. Viimeisimmässä kokeiden sarjassa he käyttivät sammakoiden eristettyjä sydänkorvakkeita.” (4)
Tulokset ovat kiistämättömiä. He saivat selville, että RNA-DNA (ribonukleiini-deoxyribonukleiini) hybridisaatiolla on suuri suhteellinen osuus samalta lajilta kerätyn bakteeri-DNA:n ja titratun sydänkorvakkeista kerätyn RNA:n välillä. (DNA, joka on kaikkien solujen tumassa oleva ominaisnukleiinihappo, on olennainen molekyyli, joka kantaa mukanaan kehon solujen geneettisen koodin.)
“Koska me tiedämme, että mikään bakteeri ei ollut päässyt sammakoiden sydänkorvakkeisiin, me voimme ainoastaan todeta että bakteeri-DNA:n on ollut erityttävä bakteereista ja eläinsolut olivat ne absorboineet”, Stroun sanoo.“Tämä siirtymäilmiö, tai transkensio, kuten tri. Anker sitä nimittää, on erittäin todennäköisesti yleinen, muutoin hän ja tri. Stroun eivät olisi todennäköisesti onnistuneet siinä ensi yrittämällä, että eläinkudokset syntetisoivat bakteeri-RNA:ta…” “Tämän työn seuraamukset transkeesiolle ovat jättiläismäiset, sillä genevalaisten työ vihjaa, että ilmiö on käynnissä koko ajan — jopa omissa kehoissamme… Voisiko esimerkiksi reumaattisen kuumeen johdosta seuraava sydänvaurio ja samanlainen bakteeri-infektio olla seurausta reumaattisesta kuumeesta ja samanlaiset bakteeri-infektiot voivat olla seurausta kehon immuunijärjestelmän reaktiosta siihen, että omat solut tuottavat vierasta RNA:ta?”
Ankerin ja Strounin myöhemmät tutkimukset vahvistivat raportin havainnot. (5)
Geneettinen hybridisaatio
Puhtaan geneettisenä materiaalina olisi odotettavissa, että virukset ovat alttiimpia geenihyppäysprosessille kuin muut mikro-organismit. Seuraava julkaisu tukee tätä hypoteesia: tuman polyhedroosiviruksen 24 näytteen tutkimuksessa niiden viljelmissä oli viruksen palasten sekä lisäyksiä että poistoja, mikä puhuisi sen puolesta että virus sekä on antanut geneettistä materiaalia soluun että vastaanottanut geneettistä materiaalia solulta, jossa se on viljelty, mikä näin myös viittaisi siihen mahdollisuuteen, että samanlaista virusvaihdantaa tapahtuu ihmisillä (oma tulkinta). (6)
Toinen mahdollinen virusinfektioiden (oletettavasti myös virusrokotteiden) komplikaatio on, että tiettyjen virusproteiinien ja aivojen ja hermojen myeliinikudosten välillä on löydetty samankaltaisuuksia. (7) Tämän virusproteiinien ja hermojärjestelmän homologisten alueiden välisen samankaltaisuuden tuloksena immunologiset ristireaktiot voivat johtaa infektion tai rokotteen jälkeiseen enkefaliittiin, myeliittiin tai neuriittiin. Näihin viruksiin kuuluu tuhkarokko, Epstein-Barr, A- ja B-influenssa ja muut hengitystieinfektioita aiheuttavat virukset.
Tätä ajatusta kehitelläksemme, artikkelissa otsikolla “Vaccination and autoimmunity-‘vaccinosis’: a dangerous liaison?”, kirjoittajat huomauttavat rokotteiden “molekyylimimiikan” ongelmasta, jossa rakenteellinen samankaltaisuus virus-antigeenin ja oman itsen antigeenin välillä saattaa, kun aiheutetaan pieni modifikaatio antigeenin kudokseen, saada sen näyttämään vieraalta immuunijärjestelmälle ja näin se voisi olla vasta-ainetuotannon kohde” (myös autoimmuniteetin). (8)
Endogeeniset ja eksogeeniset hyökkäykset immuunijärjestelmää vastaan
Palataksemme keskiaikaisen linnan analogiaan, ihmiskeho voi sietää suuret määrät myrkkyjä, mutta kun ulkoiset puolustukset otetaan pois ja geenit jäävät suojaamatta, juuri tämäntyyppisessä skenaariossa voi esiintyä teoreettisesti geenivaurioita. Tilanteet joissa tällaiseen geneettiseen haavoittuvaisuuteen ajaudutaan ovat seuraavanlaisia:
• Konferenssissa vuosia sitten tri. H.H. Fudenberg, satoja tutkimuksia julkaissut maailmankuulu immunologi, lausui seuraavasti: “Yksi rokote vähentää soluvälitteistä immuniteettia 50%:lla, kaksi rokotetta 70%:lla… kolmoisrokotteet (MMR, DTaP) merkittävästi vahingoittavat soluvälitteistä immuniteettia, mikä altistaa toistuville virusinfektioille, erityisesti korvatulehduksille, sekä hiiva- ja sieni-infektioille.”
• Vakava ja/tai pitkittynyt stressi nostaa sekä endogeenista adrenaliinia että seerumin kortisonitasoja. On pitkään tiedetty, että kortisonilääkitykset tukahduttavat immuunijärjestlemää. Endogeeniset korkeat kortisonitasot voivat tehdä samoin.
• Myrkylliset kemikaalit kuten Persianlahden sodan syndroomassa (9) tai toksisten myrkkyjen kaatopaikat, joihin on yhdistetty synnynnäiset kromosomaaliset anomaliat näillä alueilla asuvilla asukkailla. (10)
• Ravinnevajeet, erityisesti foolihapon puute, joka on kriittisessä roolissa kromosomien tuotannossa ja korjaamisessa. Foolihaposta kertovassa monografissa LT Sidney M Baker kertoo, että syövän esiasteen kromosomivaurioita on löydetty soluviljelmistä, joiden viljelmänesteessä on vähän foolihappoa. Tupakoitsijoiden veren foolihappotasot ovat matalammat ja heillä on enemmän syövän esiasteen kromosomimuutoksia kuin ei-tupakoitsijoilla. (11)
• Kuten lastentautien peruskirjoissa kerrotaan, vauvat ja lapset, joilla ei ole paljoakaan immuniteettia, ovat suurelta osin riippuvaisia äidiltään saamista vasta-aineista noin ensimmäiset 6 kk syntymästä, sillä heidän imusolmukkeensa ovat pienet, heill�� ei ole niin paljoa plasmasoluja luuytimessään eikä heidän immunoglobuliinituotantonsa ole suuri. Normaalisti arviolta 6v ikäsillä on tarpeeksi immuuniparametreja. Ainakin teoriassa, sillä immuunijärjestelmän epäkypsyys lapsuudessa tarkoittaa, että lapsen genetiikka on haavoittuvainen.
• Vaikka lopullinen näyttö on vajavaista, on paljon epäsuoraa näyttöä siitä että rokotteet saattavat vääristää ihmisen immuunijärjestelmää pois soluvälitteisestä immuniteetista, mikä on normaalisti terveellä ihmisellä dominantti, kohti heikempää vasta-aineimmuniteettia, mikä on yhteydessä allergioihin ja autoimmuunisairauksiin sekä kasvaneeseen haavoittuvaisuuteen viruksille ja sieni-infektioille. Tämä johtopäätös jää tuskin tekemättä koska suurin osa tällä hetkellä käytössä olevista ellei kaikki lapsuusajan rokotteet injektoidaan suoraan kehoon ja suunnataan vasta-aineiden tuotantoon luuytimessä. Ohittamalla limakalvot kehossa soluvälitteinen immuunijärjestelmä pysyy heikkona ja suhteellisen kitukasvuisena johtuen stimulaation puutteesta. Kuten aiemmin todettua, kun vasta-ainesysteemi muuttuu dominantiksi, niinkuin tutkimuksessa osoitettiin, dominanssi pyrkii pysymään.
Kummallakin systeemillä on identifioivat tuntomerkkinsä nimeltä sytokiinit (peptidejä jotka toimivat viestinvälittäjinä) ja tämä on se miten ne identifioidaan. Sudhir Guptan tutkimus 20 autistisella lapsella, sairaus joka monilla liittyy yhä useammin rokotteisiin, näytti konsistentisti vasta-ainesytokiinien määrän olevan koholla ja solusytokiinien matalalla. (12) Sen takia jos rokotteet vääristävät lasten immuunijärjestelmää tuomalla käyttöön vasta-ainevälitteisen järjestelmän erittäin haavoittuvaisena aikana elämässä, ne saattavat olla tuplasti tekemässä pahojaan geenimutaatioiden kannalta.
Stealth-virukset ja LT John Martinin työ
Stealth-virus on virus, joka saa aikaan pitkäaikaisen infektion ihmisissä usean vuoden ajaksi, eikä ihmisen immuunijärjestelmä löydä sitä johtuen sen geneettisestä sirpaloitumisesta ja geneettisten elementtien tilkkutäkkimäisyydestä. Tarina alkaa vuosia sitten, kun tri. Martin toimi FDA:n virusonkologian osaston johdossa, jossa hän löysi tuntematonta DNA:ta tuolloin valmistetusta suullisesti annettavasta poliorokotteesta. Myöhemmin hän sai tietää, että apinoiden sytomegaliavirusta (CMV) oli löydetty kaikista 11 afrikkalaisesta viherapinasta, joita oli tuotu poliorokotteen tuotantoon. (13)
FDA:lta lähdettyään tri. Martin toimi patologian professorina Etelä-Kalifornian yliopistossa. Siellä hän testasi verinäytteitä kroonista väsymysoireyhtymää, autismia ja muita neurosairauksia sairastavilta potilailta. Työ johti siihen, että hän löysi uniikkeja soluja tuhoavia viruksia, joita immuunijärjestelmä ei tunnistanut. Näitä kutsuttiin “stealth-viruksiksi”, joista jotkut selkeästi olivat peräisin apinoiden sytomegaliaviruksesta. Viruksilta puuttui tietyt geenit, joita ilmaisemalla ne saisivat aikaan immuunivasteita isännässä. (14-18)
Selitys kuuluu, että stealth-virus, joka tri. Martinin työn perusteella on peräisin CMV:n kontaminoimasta poliorokotteesta, oli muuttunut äärimmäisen herkäksi ja epävakaaksi, mahdollisesti useiden isännästä toiseen siirtymisen seurauksena kun rokotetta oltiin kehitetty. Epävakaampi virus teoreettisesti olisi paljon alttiimpi vaihtamaan materiaalia isäntänsä kanssa, josta lopulta tulisi eräänlainen geneettinen rubikin kuutio, jossa on tilkkutäkkimäisesti kaikenlaista materiaalia. Tämä tilkkutäkki pysyisi piilossa tartunnan saaneen isännän immuunijärjestelmältä.
Martin oli raportoinut apinoiden CMV -alkuperää koskevista kroonisen väsymysoireyhtymän (15) sekä lasten autismin (18) löydöksistään. Urnovitzin tekemät kromosomilöydökset veteraaneilla, jotka kärsivät Persianlahden sodan syndroomasta (20) puhuivat “monista enteroviruksen kaltaisista segmenteistä” epänormaaleissa kromosomeissa. Urnovitz huomautti myös, että käytännössä kaikki Persianlahden sodan veteraanit ovat saaneet suullisen poliorokotteen, mikä tarkoittaa että CMV-kontaminoitunut poliorokote on saattanut olla enterovirussegmenttien lähde (polio on enterovirus).
Kun ottaa huomioon tri. Martinin löydösten mahdolliset seuraamukset, on vain ihmeteltävä oliko näitä hämmentäviä havaintoja tarkoitus tutkia lisää, vai oltiinko asia jättämässä tuleville sukupolville?
Howard B. Urnovitzin työ ja The Chronic Illness Foundation
Tri. Urnovitz ja hänen kollegansa ovat tutkineet rokotteiden vaikutuksia syöpään, Persianlahden sodan syndroomaan, MS-tautiin ja AIDSiin. Urnovitz, jolla on sekä immunologian että mikrobiologian tohtorintutkinto Michiganin yliopistosta, jossa hän rokotteita tutki, oli muuttunut erääksi kaikkein äänekkäimmistä tieteentekijöistä, jotka olivat alkaneet puhua rokotteiden aiheuttamista geenimutaatioista. (19) Hänen työnsä tällä alalla on saanut selville mm. seuraavaa:
1. Kehoillamme on ulkoisten sen kohtaamien aineiden “geenimuisti”, johon kuuluu myös rokotteet.
2. On olemassa raja sille miten paljon materiaalia kehomme sietävät ennen kuin geenivaurioita esiintyy ja/tai ennen kuin ne muuttuvat kroonisiksi sairauksiksi.
3. Jokaisella on oma uniikki geneettinen kaavansa, joka vastaa eri aineeseen eri tavalla.
Vaikka Urnovitz ei kertonut enempää “geenimuistista”, hänen viittauksensa voidaan ymmärtää viittauksena siihen, että vanhemmiltamme perittyyn geneettiseen kaavaan vaikuttaa ja sitä mahdollisesti muuttaa adaptoituminen ympäristön eri vaikutteisiin elämämme aikana.
Ehkäpä Urnovitz ja kollegat tunnetaan parhaiten heidän Persianlahden sodan syndrooman (GWS) parissa tekemästä työstään, jossa he saivat selville näyttöä geenimuutoksista kromosomi 22q11.2:ssa, joka on tunnettu monista mutaatioistaan, joilla vaikuttaa olevan rooli GWS:n patogeneesissä. (20) Vieläkin silmiinpistävämpää on se miten he sekvensoivat löydöksensä, monien enteroviruksen kaltaisten segmenttien löytäminen viittaisi siihen, että tämä on voinut olla mukana aiheuttamassa muutoksia 22q11.2:een. Kuten aiemmin todettu, Persianlahden sodan veteraanit saivat rokotteen suullisena, enteroviruksen, mahdollisesti apinoiden CMV:n kontaminoiman.
Lisäksi raportin johdantokappaleessa kirjoittajat olivat listanneet kemikaalit, joille veteraanit olivat altistuneet Persianlahden sodan aikana, mm. kemiallisen sodankäynnin aineille, tutkimuskemikaaleille (esim. pyridostigmiinibromidi), organofosfaatti, karbamaatti ja muut hyönteismyrkyt, sekä myrkylliset palamistuotteet öljylähteiden ja diesel-kaasujen palaessa. Vaikka näitä ei oltu eritelty, listan mukaan ottaminen viittaa siihen, että myrkyllinen kemikaalialtistus on voinut olla kausaaliroolissa Persianlahden sodan syndroomassa ja sen mukanaan tuomissa geenimuutoksissa.
Tämän lisäksi jotkut geenisekvenssit on havaittu olevan peräisin muista, tunnistamattomista ei-ihmislähteistä. Tämä herättää kysymyksen siitä onko Urnovitzin ja John Martinin työn välillä yhteys, (14-18) suun kautta annettujen poliorokotteiden ja suun kautta tulleen polioviruksen apinoiden munuaissoluissa viljelemisen kautta, mikä näin kontribuoisi Urnovitzin raportissa mainitut ei-ihmissegmentti.
Urnovitzin (9, 20-22) tekemä työ asettaa rokotteiden aiheuttamat geenimutaatiot vakavaan valoon. Vanhempamme tarjoavat meille geneettisen koodin syntymässä, mutta tämä raaka geenimateriaali näyttää olevan muokattavissa ympäristön vaikutteista, mm. myrkyllisillä kemikaaleilla ja rokotteilla. Yllämainittuun informaatioon perustuen on sekä mahdollista että uskottavaa, että geneettisiä translokaatioita tapahtuu rokotteiden seurauksena. Se on varmastikin uskottava huoli.
Immunogenetiikka
Tieteentekijät eivät ymmärrä hyvin immuunijärjestelmämme genettiikkaa. Kutienkin monet tutkimukset esittävät vakavia implikaatioita. Yhtenä esimerkkinä MG Montinari ja hänen kollegansa tutkivat rokotteiden jälkeisiä keskushermostosairauksia (CNS) ja ihmisten leukosyyttien antigeenejä (HLA), jotka käytännössä ohentavat aivojen ja hermoston kudoksista myeliinipinnoitteen. (23)
HLA-järjestelmä on sellainen joka auttaa henkilön immuunijärjestelmää erottamaaan mitkä siitä ovat “omaa itseä” ja mitkä “ei-itseä”. Vaikka mekanismi on monimutkainen, se on järjestelmä joka alkiovaiheessa oppii tunnistamaan kehon terveen solun “omana itsenä” niin että nämä solut pysyvät koskemattomina immuunijärjestelmän etsi-ja-tuhoa-mekanismeilta, ja ne jättävät mekanismin suojaamaan kehoa ulkopuolisilta hyökkääjiltä.
Erikoishuolena on se, että HLA-järjestelmällä on myös alttiutta polymorfismiin (mutaatio), mutaatiot taas mahdollisesti johtavat itsetunnistuksen heikkenemiseen. Tämä prosessi voi olla juurisyy, tai se voi olla yksi pääsyistä autoimmuunisairauksille, joissa immuunijärjestelmä hyökkää kehon omien solujen kimppuun. HLA-järjestelmä on tässä prosessissa keskeisessä asemassa. (24) Kun HLA-järjestelmän alleelit mutatoituvat, kuten joskus virusinfektioiden, virusrokotteiden tai myrkyllisten kemikaalien aiheuttamien ympäristösairauksien tapauksessa, kehon immunogeneettinen muisti muuttuu. Antigeenin esittäminen immuunijärjestelmälle on tärkeää, ja tämän esityksen sotkeminen voi aiheuttaa sen että keho luulee normaalikudosta, kuten aivojen tai hermoston myeliini, hyökkääjäksi ja näin se hyökkää omaa itseään vastaan (autoimmuniteetti).
Montinari sai selville, että HLA:n tietyt alleelit (A3 & DR7) löytyivät potilailta useammin rokotteiden aiheuttamissa sairauksissa. Tämä viittaa sellaisten sairausten immunogeneettiseen syntyyn. Montinaria huoletti se, että rokotteiden lisäaineet kuten timerosaali aiheuttivat geneettisiä mutaatioita muuttamalla aminohappoja antigeeniproteiinien esityksessä (25-29), mikä voi olla syynä siihen että keho hämmentyy ja ryhtyy autoimmuunireaktioihin.
Lisähuolet rokotteiden aiheuttamista geenimutaatioista
Monet meistä tietävät The Human Genome Projectin, joka on yritys kartoittaa ihmisten geenien kaikki kromosomisijainnit. On tärkeää huomata, että geenien kartoittamisen tekniikka oikeasti sulauttaa ihmisen ja rotan kudoksia viljelyissä. Nämä solut, joita kutsutaan somaattisiksi ihmis-jyrsijäsoluiksi ovat itse asiassa sekä rotan että ihmisen kromosomit yhdistettynä. Ihmisten ja ei-ihmisten solujen hybridisaatio tapahtuu sekä kudosviljelmän avulla että toistuvien solusyklikierrosten avulla, joissa ihmisten kromosomeja “häviää”. Tämä mahdollistaa tieteentekijöille merkitä tiettyjä proteiineja ilmaisevia geenejä yksittäisiin ihmiskromosomeihin. (30)
Kun tiedetään, että sellainen hybridisaatio tapahtuu laboratorioprosesseissa ja se voidaan toistaa, pitää vain ihmetellä onko rokotteilla, jotka ovat kontaminoituneita ja tehty eri ihmisten, eläinten ja ei-ihmisten soluista/DNA:sta, samanlainen vaikutus ihmiskehoon.
PAlataksemme geneettisen kontaminaation aiheeseen, ja Ankerin ja Strounin työhön sekä ihmisen ja rotan solufuusioon, me tiedämme, että monet rokotteet käyttävät “kuolemattomia solulinjoja”, jotka ovat itse asiassa syöpäisen tyyppisiä soluja, joilla ei ole mitään rajaa sille miten monta kertaa ne voivat jakautua. Kaikkein yleisimmin tunnettu kudostyyppi on ihmisen diploidityyppi, joka on eristetty abortoidusta sikiöstä. On mahdollista, että nämä solut voivat hybridisoitua itsekseen. Itse asiassa, se on todennäköistä perustuen siihen mitä me tiedämme somaattisista ihmis-jyrsijäsoluista.
Lisäksi on huoli siitä, että nämä solulinjat kontaminoituvat helposti patogeeneilla ja levittävät syöpää (mutaatioita) aiheuttavaa materiaalia ihmisiin. (31-34)
Tietyt rokotteet kuten “rekombinantti”-, “aliyksikkö”- ja “puhdas-DNA”-rokotteet käyttävät geenimanipulaatiomenetelmiä tuotannossaan. Nämä tekniikat aiheuttavat huolta johtuen rokotteen ja ihmisproteiinin/DNA:n välisistä tuntemattomista reaktioista. FDA itse asiassa tunnustaa tämän huolen, jossa mutaatioita tapahtuu onkogeenien aktivaation tai kasvaimia poissa pitävien geenien poiskytkeytymisen tapauksissa, mikä aiheuttaa syöpää. Lisäksi he myöntävät, että vapaat nukleiinihapot helposti kulkeutuvat ja integroituvat solun genomiin, mikä mahdollisesti johtaa geenimutaatioihin. (35,36) Yksityiskohtaisempi ja teknisempi raportti, joka käy läpi monet rokotekontaminaatioiden aiheuttamat syövät ja geenivaikutukset huomauttaa, että jokaisen rokoteannoksen sallitaan sisältää 100,000,000 DNA-palasta, jotka ovat muita kuin virusta tai viruksella kontaminoituja osia. Me uskomme, että jokainen sallittu DNA-palanen on riski.
Tiivistelmä
Kirjeessä Science Magazinen päätoimittajalle, lokakuussa 1967, Joshua Lederberg, Stanfordin yliopiston lääketieteen laitoksen geeniosaston tutkija varoitti elävän viruksen rokotteista:
“Itse asiassa me manipuloimme biologisesti varsin suurella skaalalla eläviä viruksia massaimmunisaatiokampanjoissa… Yksinkertaiset viruspreparaatiot kuten yleisimmin käytössä tällä hetkellä olevat ovat myös haavoittuvaisia kontaminaatiolle ja väärintunnistukselle.” (38)
Suuremmassa mittakaavassa kyse rokotteiden mahdollisista geenivaikutuksista saattaa olla tieteellisen tiedon “musta aukko”. Vaikka sitä tapahtuisikin, onko meillä teknologiaa selvittää se, ja jos ei ole, onko meillä aikaa odottaa hitaita tiedeprosesseja sellaisten suhteiden selvittämiseksi? Tutkimukset Afrikasta, Englannista, Ruotsista ja Uudesta Seelannista ovat jatkuvasti näyttäneet lisääntyneiden allergiaongelmien kuten astman ja ihottuman esiintyvyyden, sekä sairauksien lisääntymisen, täysin rokotetuilla lapsilla verrattuna vajaasti tai ei ollenkaan rokotettuihin. (39-42) Vaikuttaa siltä kuin meille olisi käsittämätöntä että terveys voisi olla yksi asia ja genetiikka toinen, tai että nämä huonontuvan terveyden ilmiöt eivät toisi mukanaan vastaavia muutoksia geeneissä.
Meidän mielestämme tämä on yksi kaikkein olennaisimpia ongelmia, joita vastaamme on tullut, ja vanhemmat saavat vapaan tahdon hyväksyä tai hylätä rokotteet lapsiltaan perustuen informoituun suostumukseen. Aina kun katsoo luonnon maailmaa, löytää korjaavia prosesseja. Se on perussysteemi jolle Yhdysvaltain perustuslaki on muotoiltu ja jollaiseksi se on suunniteltu. Saman periaatteen tulsii soveltua lapsuusajan rokotteisiin. Ainoastaan tällä tavoin asiat voidaan korjata.
Lähdeviitteet
1. Robinson DS, Predominant TH2-like bronchoalveolar T-lymphocyte population in atopic asthma, New Engl J Med, Jan 30, 1992; 326: 298-304.
2. Holt PG, Sly PD, Allergic respiratory disease: strategic targets for primary prevention during childhood, Thorax, 1997; 52:1-4.
3. Immunobiology; The Immune System in Health and Disease, Charles Janeway, Paul Travers, Mark Walport. Donald Capra, Fourth Edition, Garland Publishing, New York, 1999:394-395.
4. Stroun M, Anker P, Transcription of spontaneously released bacterial deoxyribonucleic acid in frog auricles, J Bacteriology, April, 1973; Vol 114: 114-120.
5. Anker P, Stroun M, Bacterial ribonucleic acid in the frog brain after a bacterial peritoneal infection, Science, Nov. 10, 1972; 178:621-623.
6. Kumar S, Miller LK, Effects of serial passage of Autographa Californica Nuclear polyhedrosis virus in cell culture, Virus Research, 1987; 7:335-349.
7. Jahnke U, Fischer EH, Alvord EC, Sequence homology between certain viral proteins related to encephalomyelitis and neuritis, Science, July 19, 1985; 29:242-284.
8. Shoenfeld Y, Aron-Maor A, Vaccination and autoimmunity-‘vaccinosis’: a dangerous liaison?, J Autoimmun, Feb 2000, 14(1):1-10.
9. Urnovitz HB, Tuite JJ, Higashida JM et al, RNAs in the sera of Persian Gulf War Veterans Have Segments Homologous to Chromosome 22q11.2, Clinical and Laboratory Diagnostic Immunology, May, 1999; 6(3):330-335.
10. Vrijheid M, Dolk H, Armstrong L et al, Chromosomal congenital anomalies and residence near hazardous waste landfill sites, Lancet, January 26, 2002; 359:320-322.
11. Folic Acid, the Vital Nutrient that Fights Birth Defects, Cancer, and Heart Disease, Sidney M Baker, M.D., Keats Publishing, Inc., New Canaan, Connecticut, 1995.
12. Gupta S et al, Th1 and Th2-like cytokines in CD4+ and CD8+ T cells in autism, J Neuroimmunol, 1998; 85:106-109.
13. This story is related in the book, Emerging Viruses, AIDS and Ebola, by Leonard G Horowitz, D.M.D., M.A., M.P.H., Tetrahedron, Inc., Rockport, MA, 1997, pp 488-493.
14. Martin WJ, Ahmed KN, Zeng LC et al, African green monkey origin of the atypical cytopathic ‘stealth virus’ isolated from a patient with chronic fatigue syndrome, Clinical and Diagnostic Virology, 1995; 4:93-103.
15. Martin WJ, Anderson D, Stealth virus epidemic in the Mohave Valley, Pathobiology, 1997; 65:51-56.
16. Martin WJ, Genetic instability and fragmentation of a stealth viral genome, Pathobiology, 1996; 64:9-17.
17. Martin WJ, Consultation on detection of simian cytomegaloviruses in human tissue, (Presentation July 1, 1996 sponsored by the National Institute of Allergy and Infectious Disease (NIAID), held in the Solar Building, Rockville, MD).
18. Martin WJ, Stealth virus isolated from an autistic child, (letter to the editor), J Autism Developmental Disorders, 1995; 25(2):258.
19. Written testimony of Dr. Howard B Urnovitz, August 3rd, 1999, at the Committee on Government Reform and Oversight
20. Urnovitz HB, Tuite JJ, Higashida JM, Murphy WH, RNAs in the sera of Persian Gulf War veterans have segments homologous to chromosome 22q11.2, Clin Diagn Lab Immunol, May, 1999; 6(3):330-335.
21. Urnovitz HB et al, Increased sensitivity of HIV-2 antibody detection, Natural Med, 1997; 3:1258
22. Urnovitz HB, Murphy WH, Human endogenous retroviruses: nature, occurrence, and clinical implications in human disease, Clin Microbiol Rev, 1996; 9:72-99.
23. Montinari MG, Favoino B, Roberto A, Diagnostic role of immunogenetics in post-vaccine diseases of the CNS: preliminary results, Medit J Surg Med, 1996; 4(2):69-72.
24. Laurentaci G, Favoino B, Immunogenetica e malattie HLA associate, Oedalo Lilostampa Bari, 1999.
25. Migliore L, Nieri M, Evaluation of twelve potential aneuploidogenic chemicals by the in vitro human lymphocyte micronucleus assay, Toxic in Vitro, 1991; 5(4):325-336.
26. Shrana I et al, Mitosis and numerical chromosome aberration analyses in human lymphocytes: 10 known or suspected spindle poisons: Mutation Research, 1993; 287:57-70.
27. Miller BM, Adler ID, Aneuploid induction in mouse spermatocytes mutogenesis, Mutogenesis, 1992; 7(1):69-76.
28. Gudi R et al, Assessment of the in vivo aneuploidy/micronucleus assay in mouse bone marrow cells with 16 chemicals, Env Mol Mutagen, 1992; 20:106-116.
29. Ilse-Dore A, Synopsis of the in vivo results obtained with the 10 known or suspected aneugens tested in the CEC collaborative study, Mutation Research, 1993; 287:131-137.
30. Nelson Textbook of Pediatrics, 16th Edition, WB Saunders C., 2000, page 315.
31. Harasawa R, Latent Risk in Bovine Serums Used for Biopharmaceutical Production, http://www.asmusa.org/pcsrc/sumO2.htm
32. Levings RL, Wessman SJ, bovine diarrhea virus contamination of nutrient serum, cell cultures, and viral vaccines, Dev Biol Stand, 1991; 75:177-181.
33. Giangaspero M et al, Genotypes of pestivirus RNA detected in live virus vaccines for human use, J Vet Med Sci, 2001: 63(7):723-733. PMID 11503899
34. Harasawa R, Miznsawa H, Detection of Pestiviruses from Mammalian cell cultures by PCR, Procedings of 3rd Internat World Congress on Biomedical Sciences, 1996; 12.-9.-20 Riken, Isukuba, Japan, http://www.3iwc.riken.go.jp/congress/sympo/sbb0202/ako111/tit.htm
35. Ho M et al, Slipping through the regulatory net: ‘Naked’ and ‘free’ nucleic acids. TWN Biotechnology and Biosafety Series, No. 5, 2001. http://www.twnside.org.sg/title/biod5.htm
36. Points to consider on Plasmid DNA vaccines for preventive infectious disease indications. FDA/CBER, Office of Vaccine Research and Review, 1996, http://www.fda.govc/cber/glns/plasmid.txt
37. McRearden B, What is coming through that needle? The problem of pathogenic vaccine contamination, 2002, http://www.jefsutherland.org/complementary/vaccine_contamination_mcrearden.pdf
38. Lederberg J, Letter-to-the-Editor, Science, Oct. 20, 1967:313.
39. Shaneen SO et al, Measles and atopy in Guinea-Bissau, Lancet, June 19, 1996; 347:1792-1796.
40. Odent, MR, Pertussis vaccine and asthma; is there a link? JAMA, 1994; 271:229-231.
41. Alm JS et al, Atopy in children of families with anthroposophic lifestyle, Lancet, May 1, 1999; 353:1485-1488.
42. Kemp T et al, Is infant immunization a risk factor for childhood asthma or allergy?, Epidemiology, Nov., 1997; 8(6):678-680.
Artikkelin julkaissut whale.to
https://eksopolitiikka.fi/tiede/rokotteet-ja-geenimutaatio/?utm_source=TR&utm_medium=Tumblr+%230&utm_campaign=SNAP%2Bfrom%2B_%7C+Eksopolitiikka.fi+%7C_
0 notes
Text
Euroopan eliitti ja väärennetyt rokotekortit: Skandaali paljastui
Yli 2 200 julkisuuden henkilöä, virkamiestä ja Euroopan eliittiä jäi kiinni väärennetyistä rokotekorteista ja valehtelusta omista COVID-19-rokotustiedoistaan. Näiden henkilöiden joukossa oli julkkiksia, liikemaailman johtajia ja jopa lääketeollisuuden avainhenkilöitä, jotka olivat saaneet suolavesiruiskeen mRNA-rokotteiden sijaan. He maksoivat merkittäviä summia väärennetyistä rokotetiedoista kansallisessa rokotusrekisterissä.
Operation Jenner paljastaa laajan petoksen
Euroopan poliisivoimat suorittivat laajamittaisen tutkimuksen, joka tunnetaan nimellä Operation Jenner. Operaatio paljasti, että väärennettyjen rokotepassien ja -korttien hinnat vaihtelivat henkilön sosiaalisen aseman mukaan. Esimerkiksi tunnetut julkisuuden henkilöt ja Big Pharman johtajat maksoivat huomattavasti enemmän, mikä viittaa siihen, että he ymmärsivät rokotteiden mahdolliset riskit ja halusivat välttää niitä hinnalla millä hyvänsä.
Yksi merkittävimmistä nimistä oli eurooppalaisen lääkejättiläisen Pharma-Marin puheenjohtaja Jose Sousa-Faro, jota vastaan on nostettu rikossyytteitä väärän rokotuksen hankkimisesta. Tuhansien muiden henkilöiden nimet sisältyvät myös tutkintaan liittyviin asiakirjoihin.
Kaksoisstandardit rokotevaatimuksissa
Tämä tapaus on herättänyt laajaa suuttumusta ja kysymyksiä poliitikkojen, julkkisten ja eliitin rehellisyydestä. Monet heistä kampanjoivat julkisesti rokotusten puolesta ja vaativat tiukkoja rokotepassivaatimuksia, mutta yksityisesti välttivät itse rokotteet maksamalla suolavesiruiskeista ja väärennetyistä asiakirjoista.
Erityisesti herää kysymys, kuinka nämä henkilöt, jotka saarnasivat "rokotekultin" evankeliumia – väittäen, että rokotteet ovat välttämättömiä yhteiskunnan suojelemiseksi – saattoivat itse kiertää rokotuksia. Tämä kaksoisstandardi on lisännyt epäluottamusta ja vaatimuksia oikeudenmukaisuudesta.
Paralleelit Yhdysvalloissa
Tapauksen on epäilty heijastavan myös Yhdysvalloissa mahdollisesti tapahtuneita käytäntöjä. Monet uskovat, että osa poliitikoista ja julkisuuden henkilöistä on saattanut saada suolavesiruiskeita suorassa televisiolähetyksessä välttääkseen mRNA-rokotteet. Näiden henkilöiden käyttäytyminen näyttää ristiriitaiselta, kun he samalla korostivat rokotteiden turvallisuutta ja tehokkuutta.
Oikeudenmukaisuuden vaatimus
Tapaus on johtanut vaatimuksiin oikeudellisista seuraamuksista niille, jotka osallistuivat tähän petolliseen juoneen. Monet katsovat, että niiden, jotka aktiivisesti ajoivat rokotuspassivaatimuksia ja rajoituksia mutta itse kiersivät ne, tulisi kohdata oikeudellinen vastuu toiminnastaan.
Rokotekorttiskandaali on tuonut päivänvaloon valtavan petoksen, joka horjuttaa luottamusta julkisiin instituutioihin, poliittiseen johtoon ja lääketieteelliseen yhteisöön. Tapaus muistuttaa tarpeesta lisätä läpinäkyvyyttä ja rehellisyyttä sekä varmistaa, että kaikilla yhteiskunnan tasoilla noudatetaan samoja sääntöjä.
Timo Tynkkynen
0 notes
Text
Hörhöt väittävät, että rokotteet ovat myrkkyä ja jokainen rokote tappaa. Euroopassa on rokotettu 90% väestöstä, joten se tekee noin 675 miljoonaa kuollutta. Missä ruumiita säilytetään?
0 notes
Photo
Neljän COVID-rokotteen Euroopassa julkisesti saatavilla olevan datan tarkastelua (30.3.2021)

Kirjoittanut: Catherine FRADE
Julkaistu: 1. huhtikuuta 2021

Lääketieteen tohtorina, entisenä lääketeollisuuden kansainvälisen sääntelyn johtajana haluan valaista ymmärrystänne virallisista tiedoista koskien neljää COVID-19 -rokotetta. Kaikki alla esitetyt tiedot on ladattavissa tästä linkistä. Olin innostunut työstäni laillisten tekstien kenttätutkimuksessa, jotta osaisin kehittää parhaiten lääkkeitä ja työskennellä synergiassa yrityksen ja terveysviranomaisten eri osastojen kanssa Ranskassa ja ulkomailla (jopa 83 maahan). 30. maaliskuuta 2021 Pfizer-, Moderna-, Astra Zeneca- ja Janssen-laboratoriot ovat saaneet ehdollisen myyntiluvan 4: lle COVID-19-rokotteelle (joulukuun 2020 ja maaliskuun 2021 välillä). Nykytilanteen ymmärtämiseksi on välttämätöntä käyttää lähdetietoja, kentällä toimimattoman henkilön on vaikea tunnistaa. Tämän artikkelin tarkoituksena on siis valaista julkista tietoa näistä neljästä COVID-19-rokotteesta viittaamalla kuhunkin lähteeseen, jotta niiden aitous voidaan vahvistaa. Tästä syystä löydät tästä artikkelista linkit myyntilupien virallisiin asiakirjoihin ja yhteenvedot Euroopan lääkeviraston (EMA) arvioimista tieteellisistä tutkimuksista .
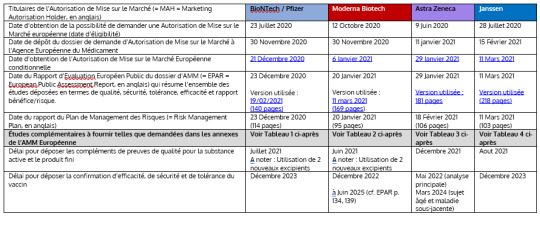
Ensinnäkin haluan selventää, että minulla ei ole eturistiriitoja lääketeollisuuden kanssa.
1. Ensinnäkin on tärkeää tietää, mikä ehdollinen myyntilupa on:
Myyntilupa (Marketing Authorization, MA/ML) myönnetään, kun tuote on osoittanut laadun, tehokkuuden ja turvallisuuden positiivisella hyöty-riskisuhteella (eli että sillä on enemmän hyötyjä kuin riskejä). Myyntiluvan saaminen on edellytys lääkkeen, mukaan lukien rokotteet, myymiselle. Kaikki MA-hakemuksen aikana tehdyt tutkimukset on tiivistetty EPAR-lausekkeeseen (= European Public Assessment Report). Se julkaistaan Euroopan lääkeviraston (EMA) verkkosivustolla. Mukana ovat myös suunnitellut tutkimukset, joita ei ole vielä tehty. Tämä aikataulu, joka kestää vuodet 2021-2025 COVID-19-rokotteesta riippuen, on määritelty ehdollisen myyntiluvan liitteissä ja EPAR-lausunnossa. Myyntilupa myönnetään myyntiluvan haltijalle (Marketing Authorization Holder, MAH). Ehdolliset myyntiluvat saatiin laadun, kliinisen ja ei-kliinisen tiedon perusteella rokotetestistä ja/tai bibliografisesta kirjallisuudesta. Bibliografisen kirjallisuuden tiedot ovat tunnustetuissa tieteellisissä lehdissä julkaistuja tutkimuksia, jotka on kirjoittanut myyntilupaa pyytäneen laboratorioon liittymättömät ja ulkopuoliset ryhmät. Nopeutetulla keskitetyllä menettelyllä saatu eurooppalainen myyntilupa sallii samanaikaisen markkinoinnin seuraavissa 30 maassa (Euroopan unioni ja Euroopan vapaakauppaliitto): Saksa, Itävalta, Belgia, Bulgaria, Kypros, Kroatia, Tanska, Espanja, Viro, Suomi, Ranska, Kreikka, Unkari, Irlanti, Islanti, Italia, Latvia, Liechtenstein, Liettua, Luxemburg, Malta, Norja, Alankomaat, Puola, Portugali, Romania, Slovakia, Slovenia, Ruotsi, Tšekki. Esimerkiksi Pfizerin EPAR (sivut 67 ja 114) päivämäärällä 19. helmikuuta 2021, joka on tähän mennessä eniten käytetty COVID-19-rokote, jolla on laajin käyttöindikaatio (yli 16-vuotiaat), mainitsee että tärkein ns. kliininen tutkimus on vaiheen 1/2/3 tutkimus, joka on edelleen kesken.
Alkuperäinen englanninkielinen EPAR Käännös Study C4951001: A Phase 1/2/3, Placebo-Controlled, Randomized, Observer-Blind, Dose-Finding Study to Evaluate the Safety, Tolerability, Immunogenicity, and Efficacy of SARS-COV-2 RNA Vaccine Candidates Against COVID-19 in Healthy Individuals The safety evaluation is based on one ongoing Phase 2/3 study that at the time of data cut-off (14-Nov-20) included 43,448 subjects who received either two doses of BNT162b2 30μg (n = 21,720) or placebo (n = 21,728). Overall, the reported reactogenicity profile are in line with any authorized vaccine. In addition, the frequency of reported AEs and SAEs were low. The emerging safety profile is presently considered favorable. Long term safety data, interaction with other vaccines, data on use in pregnancy and other subgroups (eg frail subjects, or subjects with pre-existing autoimmune diseases) are missing at this stage. The lack of long-term follow up renders the data provided non-comprehensive. Therefore, the delivery of the final C4951001 study report, including a 2-year follow up of the studied population, is classified as a specific obligation in the context of a conditional marketing authorization. The plan for the generation of further safety data post authorization is described in the section below. Tutkimus C4951001: Vaihe 1/2/3, lumekontrolloitu, satunnaistettu, tarkkailija-sokea, annosmääritystutkimus rokottajaehdokkaiden turvallisuuden, siedettävyyden, immunogeenisuuden ja tehokkuuden arvioimiseksi SARS-COV-2-RNA-rokotteelle COVID-19:a vastaan terveillä yksilöillä. (sivu 67) Turvallisuusarviointi perustuu käynnissä olevaan vaiheen 2/3 tutkimukseen, johon tietojen sulkemishetkellä (14.-20.11.) osallistui 43 448 potilasta, jotka saivat joko kaksi 30 μg: n annosta (n = 21 720) tai lumelääkettä (n = 21 728). Ilmoitettu reaktogeenisuusprofiili on kaiken kaikkiaan yhdenmukainen minkä tahansa lisensoidun rokotteen kanssa. Lisäksi ilmoitettujen haittavaikutusten ja vakavien haittavaikutusten esiintymistiheys oli pieni. Uusia turvallisuusprofiileja pidetään tällä hetkellä suotuisina. Pitkäaikaiset turvallisuustiedot, vuorovaikutus muiden rokotteiden kanssa, tiedot käytöstä raskauden aikana ja muut alaryhmät (esimerkiksi heikot tai koehenkilöt, joilla on jo autoimmuunisairauksia) puuttuvat tässä vaiheessa. Pitkän aikavälin seurannan puuttuminen tekee toimitetuista tiedoista tyhjentäviä. Siksi tutkimuksen C4951001 loppuraportin toimittaminen, mukaan lukien tutkimusväestön kahden vuoden seuranta, luokitellaan erityiseksi velvoitteeksi ehdollisen myyntiluvan yhteydessä. Suunnitelma ylimääräisten suojaustietojen tuottamiseksi valtuutuksen jälkeen on kuvattu alla olevassa osassa.
2. Yhteenvetotaulukko EMA:n verkkosivustolla julkaistusta neljän COVID-19-rokotteen myyntiluvasta:
Rekisteröintimenettelyn etenemisestä ja vielä toimitettavista tutkimuksista on oltava näkyvyys. Alla olevassa taulukossa on esitetty viralliset julkiset tiedot EMA-sivustolta: Nämä tiedot on otettu virallisista teksteistä 22. maaliskuuta 2021 (Alleviivatut tekstit viittaavat vastaaviin linkkeihin EMA:n virallisilla verkkosivuilla ja kaikki linkit on tiivistetty tämän asiakirjan loppuun).
Myyntiluvan haltijat (= MAH) BioNTech / Pfizer Moderna Biotech Astra Zeneca Janssen Päivämäärä, jolloin saatiin mahdollisuus hakea eurooppalaista myyntilupaa (kelpoisuuspäivä) 23. heinäkuuta 2020 12. lokakuuta 2020 9. kesäkuuta 2020 28. heinäkuuta 2020 Myyntilupahakemuksen jättöpäivä Euroopan lääkevirastolle 30. marraskuuta 2020 30. marraskuuta 2020 11. tammikuuta 2021 15. helmikuuta 2021 Ehdollisen eurooppalaisen myyntiluvan saamispäivä 21. joulukuuta 2020 6. tammikuuta 2021 29. tammikuuta 2021 11. maaliskuuta 2021 Päiväys, jona myyntiluvan haltijoiden asiakirja-aineistoa koskeva eurooppalainen julkinen arviointiraportti (= EPAR = englanniksi European Public Assessment Report) sisältää yhteenvedon kaikista toimitetuista tutkimuksista laadun, turvallisuuden, suvaitsevaisuuden, tehon ja hyöty-riskisuhteen suhteen. 23. joulukuuta 2020 Käytetty versio: 19.02.2021 (140 sivua) 20. tammikuuta 2021 Käytetty versio: 11. maaliskuuta 2021 (169 sivua ) 29. tammikuuta 2021 Käytetty versio: 181 sivua 11. maaliskuuta 2021 Käytetty versio (218 sivua) Riskienhallintasuunnitelman raportin päivämäärä 23. joulukuuta 2020 (114 sivua) 20. tammikuuta 2021 (95 sivua) 18. helmikuuta 2021 (106 sivua) 11. maaliskuuta 2021 (103 sivua) Lisätutkimukset toimitetaan Euroopan myyntiluvan liitteiden vaatimusten mukaisesti Katso alla oleva taulukko 1 Katso alla oleva taulukko 2 Katso alla oleva taulukko 3 Katso alla oleva taulukko 4 Määräaika tehoaineen ja lopputuotteen laatua koskevan lisätodistuksen toimittamiseksi Heinäkuu 2021 Huomaa: 2 uuden apuaineen käyttö Kesäkuu 2021 Huomaa: 2 uuden apuaineen käyttö Joulukuu 2021 Elokuu 2021 Määräaika rokotteen tehokkuuden, turvallisuuden ja sietokyvyn vahvistamisen toimittamiselle Joulukuu 2023 Joulukuu 2022 kesäkuuhun 2025 (vrt. EPAR s. 134, 139) Toukokuu 2022 (pääanalyysi) Maaliskuu 2024 (vanhukset ja perussairaus) Joulukuu 2023
3. Näiden julkisten tietojen analyysistä käy ilmi, että:
Nämä rokotteet ovat saaneet ehdollisen myyntiluvan, joka on voimassa yhden vuoden tavallisten myyntilupien viiden vuoden sijaan. Käynnissä olevat ja suunnitellut tutkimukset on saatava päätökseen, jotta voidaan hankkia vakiomuotoinen MA.
Vaikka kliiniset tutkimukset on suunniteltu alkavaksi, ne eivät ole täydellisiä, ja jotkut eivät ole vielä alkaneet. Rokotteesta riippuen lopulliset määräajat suunnitellaan vuosien 2022 ja 2025 välille (katso yllä oleva taulukko).
Yhden laboratorion COVID-19- rokotteen vaihdettavuudesta muiden laboratorioiden muiden COVID-19-rokotteiden kanssa rokotusaikataulun täyttämiseksi ei ole tietoa.
Käyttö rokote on tarkoitettu 18-vuotiaille , paitsi että Pfizerin ilmoitettu ikä on 16-vuotiaasta.
”Rokotteiden turvallisuutta ja tehoa lapsilla ja alle 18-vuotiailla nuorilla ei ole vielä varmistettu.” Tämä koskee Modernaa, Astra-Zenecaa ja Janssenia, joista ” tietoja ei ole saatavilla”. Sama Pfizerin tapauksessa ”lapsilla ja alle 16-vuotiailla nuorilla, joiden tietoja on rajoitetusti”.
Tietoa raskaana olevista naisista on hyvin rajallisesti (hylkäämiskriteerejä kliinisistä tutkimuksista): vähän tai ei lainkaan tietoa turvallisuudesta ja tehosta on toistaiseksi tiedossa. ( Taulukko 5 esimerkkinä). Raskaana olevien naisten rokottamista voidaan harkita vain tapauskohtaisesti. Pfizer-, Moderna- ja Janssen-rokotteiden osalta myyntiluvan liitteessä I esitetyssä tieteellisessä esitteessä (valmisteyhteenveto) ilmoitetaan, että Rokotteen käytöstä raskaana oleville naisille on vain vähän tietoja. Eläinkokeet eivät ole tuottaneet suoria tai epäsuoria haitallisia vaikutuksia raskauteen, alkion/sikiön kehitykseen, synnytykseen tai postnataaliseen kehitykseen (ks. AMM: n liitteessä I oleva 5.3 kohta). Käyttöä raskaana oleville naisille tulee harkita vain, jos mahdolliset hyödyt ovat suuremmat kuin mahdolliset riskit äidille ja sikiölle . ” Astra Zenecan pakkausselosteessa ei mainita vastaavia tietoja: ”Jos olet raskaana tai imetät, epäilet olevasi raskaana tai suunnittelet lapsen hankkimista, kysy neuvoa lääkäriltäsi, apteekista tai sairaanhoitajalta rokotteen ottamiseksi.”
Kokemuksen perusteella mikä tahansa markkinoille saatettu lääke antaa mahdollisuuden tuoda esiin laajamittaisia haittavaikutuksia, joita ei ole esiintynyt tai on harvoin esiintynyt kliinisissä tutkimuksissa. Tämä tarkoittaa konkreettisesti, että rokotuksiin liittyvät haittavaikutukset voivat ilmetä ajan myötä (mikä todennäköisesti selittää Astra-Zeneca-jakson maaliskuun puolivälissä 2021).
Nopeutetun keskitetyn eurooppalaisen menettelyn mukainen rokotteiden ehdollinen myyntilupa ”annettiin kansanterveyden eduksi täyttämään lääketieteellinen tarve”.
Jotkut professorit ja lääkärit ilmoittivat havainneensa alalla lääkeyhdistelmistä muodostettujen hoitojen tehokkuuden: viruslääkkeet, antibiootit, vitamiinit, ravintolisät jne. Tiedämme sen jälkeen tieteellisten julkaisujen erilaisesta kohtelusta käydyn keskustelun, joka voisi olla oikeudellisesti hyväksyttävää uusien rokotteiden ehdollisen myyntilupahakemuksen yhteydessä ja jota ei voida hyväksyä monien vuosien ajan käytettyjen lääkkeiden käytöstä (vrt. hydroksiklorokiini) …Jopa nykypäivänä asiasta edelleen debatoidaan.
Johtopäätös
Yhteenvetona voidaan todeta, että 30 maassa epätäydellisten ja/tai bibliografisten tutkimusten ja tulevien tutkimusten perusteella saatu ehdollinen eurooppalainen myyntilupa antaa ymmärtää, miten COVID-19-rokotteen antaminen vuonna 2021 on laajamittaisesti tutkittu. Ihmiset, jotka on rokotettu osana vielä kesken olevia tai tulevia tutkimuksia (kuten lapset, raskaana olevat naiset ja kaikki EPAR-taulukoissa esitetyt taulukot), joutuvat siksi kokeellisten tutkimus- ja kehitysprotokollien alaisiksi. Jokaisella on ennen rokotusta oikeus pyytää kaikki tietoisen suostumuksensa kannalta hyödylliset tiedot, mukaan lukien lääkityksen ohjeet (katso myyntiluvan liitteet, linkit tämän asiakirjan lopussa olevaan liitteeseen 6. Asiakirjat). Lisäksi ennalta varautumisen periaatteen on oltava vallitseva jo ennen mainitun suostumuksen saamista.

Tänään olen suuntautunut ammatillisessa toiminnassani ihmisten ja organisaatioiden terveyden ja elämänlaadun tukemiseen ja kouluttamiseen heidän tarkoituksensa mukaisesti, ja olen hyvin kiintynyt kaikkien kunnioittamiseen. Farmasian tohtorina ja kokonaisvaltaisena työpsykologina (CV liitteenä) yhdistän rationaaliset tieteet tietoisuuden tulokulmiin kaikissa muodoissaan. Näiden dokumentti- ja sääntelylähteiden tuella halusin antaa sinun löytää itsellesi hyvän ymmärryksen teksteistä ja siten valaista näitä uusia rokotteita. Dr. Catherine FRADE, ansioluettelo löytyy tästä. Farmasian tohtori, psykologi ja työpsykopatologi
Taulukko 1: Pfizer-myyntiluvan liite IIE, sivut 18 ja 19

Taulukko 2: Modernan myyntiluvan liite IIE, sivu 15
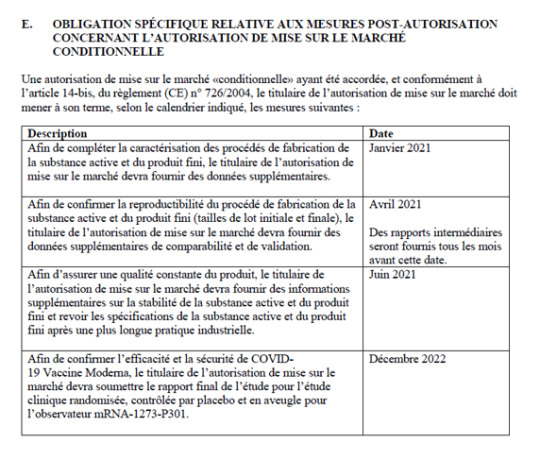
Taulukko 3: Astra Zenecan myyntiluvan liite IIE, sivut 14 ja 15
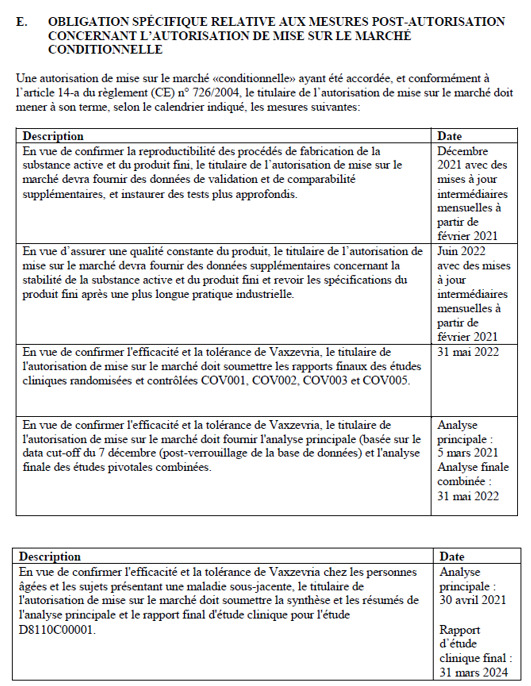
Taulukko 4: Janssenin myyntiluvan liite IIE, sivu 18
Taulukko 5: Raskaustiedot Pfizer EPAR:sta, esimerkkinä
Termi ”raskaana” tai ”raskaus” esiintyi EMA: n julkaisemassa EPAR-raportissa 10 sivulla . Raportin tarkat lauseet näkyvät sellaisina kuin ne ovat tässä taulukossa.
Extracts from the Pfizer EPAR – original text in English French translation by Deepl software Page 14. A study in pregnant women is also planned in the EU. A Post-Approval Active Surveillance Safety Study to Monitor Real-World Safety of Comirnaty (Study C4591010) will be conducted in the EU using primary data collection that monitors a cohort of vaccinees and evaluates risk of AESIs (adverse event of special interest) Sivu 14. Raskaana oleville naisille on suunnitteilla tutkimus myös EU: ssa. Hyväksynnän jälkeinen aktiivisen valvonnan turvallisuustutkimus Comirnatyn todellisen turvallisuuden seuraamiseksi (tutkimus C4591010) suoritetaan EU:ssa ensisijaisen tiedonkeruun avulla, joka seuraa rokotettujen kohorttia ja arvioi AESI-tautien (erityistä kiinnostusta aiheuttavan haittatapahtuman) riskiä. Page 68. 2.5.2. Main study, Title of study: Study C4951001: A Phase 1/2/3, Placebo-Controlled, Randomized, Observer-Blind, Dose-Finding Study to Evaluate the Safety, Tolerability, Immunogenicity, and Efficacy of SARS-COV-2 RNA Vaccine Candidates Against COVID-19 in Healthy Individuals Methods: Study Participants – exclusion criteria – Women who are pregnant or breastfeeding. Sivu 68. 2.5.2. Päätutkimus, tutkimuksen nimi: Tutkimus C4951001: Vaihe 1/2/3, lumelääkekontrolloitu, satunnaistettu, tarkkailija-sokea, annoshakututkimus SARS-COV-2-RNA-rokotteidenn turvallisuuden, siedettävyyden, immunogeenisuuden ja tehokkuuden arvioimiseksi terveillä henkilöillä COVID-19:a vastaan Menetelmät: Tutkimuksen osallistujat – poissulkemisperusteet – Raskaana olevat tai imettävät naiset. Page 93. Immunocompromised subjects and pregnant or breastfeeding women were excluded from the study. Sivu 93. Immuunipuutteiset potilaat ja raskaana olevat tai imettävät naiset suljettiin pois tutkimuksesta. Page 109. At the time of the data cut-off in the Phase 2/3 study (14 Nov 2020), a total of 23 participants had reported pregnancies in the safety database, including 9 participants who withdrew from the vaccination period of the study due to pregnancy. These participants are being followed for pregnancy outcomes. Thus, data on pregnancy are very limited at this stage. Sivu 109. Vaiheen 2/3 tutkimuksen tietojen katkaisuhetkellä (14. marraskuuta 2020) yhteensä 23 osallistujaa ilmoitti raskauksista turvallisuustietokannassa, mukaan lukien 9 osallistujaa, jotka vetäytyivät tutkimuksen rokotusjaksosta. raskauden vuoksi. Näitä osallistujia seurataan raskaustulosten suhteen. Siksi tiedot raskaudesta ovat tässä vaiheessa hyvin rajalliset. Page 113. 23 participants reported pregnancies in the safety database, nine of them were withdrawn from the study due to the pregnancy status. These participants will be followed up for pregnancy outcomes. Sivu 113. 23 osallistujaa ilmoitti raskauksista turvallisuustietokannassa, joista yhdeksän jätettiin pois tutkimuksesta raskaustilan vuoksi. Näitä osallistujia seurataan raskaustulosten suhteen. Page 114. clinical safety: Long term safety data, interaction with other vaccines, data on use in pregnancy and other subgroups (eg frail subjects, or subjects with pre-existing autoimmune diseases) are missing at this stage. Sivu 114. kliininen turvallisuus: Pitkäaikaiset turvallisuustiedot, yhteisvaikutus muiden rokotteiden kanssa, tiedot raskauden ja muiden alaryhmien käytöstä (esim. heikot tai koehenkilöt, joilla on jo autoimmuunisairauksia) puuttuvat tässä vaiheessa. Page 115. safety information: missing information = Use during pregnancy and while breast feeding Sivu 115. Turvallisuustiedot: puuttuvat tiedot = Käyttö raskauden ja imetyksen aikana Page 119. atypical COVID-19 in a cohort of people within the Department of Defense Healthcare System. For December 2023 Sivu 119. epätyypillinen COVID-19 puolustusministeriön terveydenhuoltojärjestelmän ihmisten joukossa. Joulukuussa 2023 Page 120. Study ( study short name, and title) Status (planned / on-going) : C4591015 Planned Summary of Objectives Planned clinical study to assess safety and immunogenicity in pregnant women who receive COVID-19 mRNA vaccine. Safety and immunogenicity of COVID-19 mRNA vaccine in pregnant women Safety concerns addressed: Use in pregnancy and while breast feeding. Protocol draft submission: 28-Feb-2021 Final CSR (Clinical study report) submission: 30-Apr-2023 Sivu 120. Tutkimus (tutkimuksen lyhyt nimi ja nimi) Status (suunniteltu / meneillään): C4591015 – tutkimus suunniteltu (ei vielä suoritettu) Yhteenveto tavoitteista: Suunniteltu kliininen tutkimus turvallisuuden ja immunogeenisuuden arvioimiseksi raskaana oleville naisille, jotka saavat COVID-19-mRNA-rokotetta. COVID-19-mRNA-rokotteen turvallisuus ja immunogeenisuus raskaana oleville naisille. Käsiteltyjä turvallisuuskysymyksiä: Käyttö raskauden ja imetyksen aikana. Protokollaluonnoksen toimittaminen: 28. helmikuuta-2021 Kliinisen tutkimusraportin lopullinen toimittaminen : 30. huhtikuuta-2023 Page 120. Study ( study short name, and title) Status (planned / on-going) : ACCESS / VAC4EU Planned Summary of Objectives Assessment of occurrence of safety events of interest, including severe or atypical COVID-19 in real-world use of COVID-19 mRNA vaccine. Safety concerns addressed: Anaphylaxis AESI-based safety events of interest including vaccine associated enhanced disease Use in pregnancy Use in immunocompromised patients Use in frail patients with co-morbidities (eg, chronic obstructive pulmonary disease (COPD), diabetes, chronic neurological disease, cardiovascular disorders) Use in patients with autoimmune or inflammatory disorders Protocol draft submission: 28-Feb-2021 Final CSR submission: 31-Jan-2024 Sivu 120. Tutkimus (tutkimuksen lyhyt nimi ja nimi) Status (suunniteltu / käynnissä): ACCESS / VAC4EU suunniteltu (ei vielä toteutettu) Yhteenveto tavoitteista Arvio kiinnostavien turvallisuustapahtumien esiintymisestä, mukaan lukien vaikea tai epätyypillinen COVID-19 COVID-19-mRNA-rokotteen tosiasiallisessa käytössä. Käsitellyt turvallisuuskysymykset: Anafylaksia Kiinnostavat turvallisuustapahtumat, jotka perustuvat erityisen kiinnostaviin haittatapahtumiin, mukaan lukien rokotteeseen liittyvät lisääntyneet sairaudet. Käytä raskauden aikana Käyttö immuunipuutteisilla potilailla Käyttö heikoilla potilailla, joilla on samanaikaisia sairauksia (esim. Krooninen obstruktiivinen keuhkosairaus (COPD), diabetes, krooninen neurologinen sairaus, sydän- ja verisuonisairaudet) Käyttö potilaille, joilla on autoimmuuni- tai tulehdussairaus. Pöytäkirjaluonnoksen toimittaminen : 28. helmikuuta 2021 Kliinisen tutkimusraportin lopullinen toimittaminen : 31. tammikuuta-2024 Page 136. Benefit-risk assessment and discussion 3.7.1. Importance of favorable and unfavorable effects There are no data on use in pregnant women, but a protective effect is anticipated. In the light of the reassuring data from the DART study, noting that pregnancy as such is a risk factor for severe COVID-19, and that pregnant women may additionally belong to other risk groups, vaccination may be considered on a case by case basis. Sivu 136. Hyöty-riskiarviointi ja keskustelu 3.7.1. Suotuisien ja epäedullisten vaikutusten merkitys Ei ole tietoa käytöstä raskaana oleville naisille, mutta suojaavan vaikutuksen odotetaan olevan. DART-tutkimuksen rauhoittavien tietojen valossa, jossa todetaan, että raskaus sinänsä on vakavan COVID-19:n riskitekijä ja että raskaana olevat naiset voivat lisäksi kuulua muihin riskiryhmiin, rokotuksia voidaan harkita tapauskohtaisesti.
Liite 6. Internetissä saatavilla olevat viitteet (ei tyhjentävä luettelo)
Aiheesta on olemassa suuri määrä erittäin mielenkiintoisia asiakirjoja:
Euroopan lääkevirasto (EMA): https://europa.eu/european-union/about-eu/agencies/ema_fr
Kansallinen lääkkeiden ja terveystuotteiden turvallisuusvirasto (ANSM): https://ansm.sante.fr/qui-sommes-nous/
Kysymykset ja vastaukset: Ehdollinen myyntilupa COVID-19-rokotteille EU:ssa: https://ec.europa.eu/commission/presscorner/detail/en/QANDA_20_2390
https://www.ema.europa.eu/en/news/meeting-highlights-pharmacovigilance-risk-assessment-committee-prac-8-11-march-2021
Asiakirjat Pfizer Moderna AstraZeneca Janssen Tiedot ovat saatavilla ilmaiseksi EMA: n kautta https://www.ema.europa.eu/en/medicines/human/EPAR/comirnaty https://www.ema.europa.eu/en/medicines/human/EPAR/covid-19-vaccine-moderna https://www.ema.europa.eu/en/medicines/human/EPAR/covid-19-vaccine-astrazeneca https://www.ema.europa.eu/en/medicines/human/EPAR/covid-19-vaccine-janssen Myyntiluvan liitteet https://www.ema.europa.eu/en/documents/product-information/comirnaty-epar-product-information_fr.pdf https://www.ema.europa.eu/fi/documents/product-information/covid-19-vaccine-moderna-epar-product-information_fr.pdf https://www.ema.europa.eu/fi/documents/product-information/vaxzevria-previously-covid-19-vaccine-astrazeneca-epar-product-information_fr.pdf https://www.ema.europa.eu/fi/documents/product-information/covid-19-vaccine-janssen-epar-product-information_fr.pdf EPAR-arviointikertomus – lääkärintarkastus https://www.ema.europa.eu/fi/documents/assessment-report/comirnaty-epar-public-assessment-report_en.pdf https://www.ema.europa.eu/fi/documents/assessment-report/covid-19-vaccine-moderna-epar-public-assessment-report_en.pdf https://www.ema.europa.eu/en/documents/overview/covid-19-vaccine-astrazeneca-epar-medicine-overview_fr.pdf https://www.ema.europa.eu/fi/documents/assessment-report/covid-19-vaccine-janssen-epar-public-assessment-report_en.pdf Yhteenveto lääkevalmistekomitean myönteisestä lausunnosta https://www.ema.europa.eu/en/documents/smop-initial/chmp-summary-positive-opinion-comirnaty_en.pdf https://www.ema.europa.eu/fi/documents/smop-initial/chmp-summary-positive-opinion-covid-19-vaccine-moderna_en.pdf https://www.ema.europa.eu/fi/documents/smop-initial/chmp-summary-positive-opinion-covid-19-vaccine-astrazeneca_en.pdf https://www.ema.europa.eu/fi/documents/smop-initial/chmp-summary-positive-opinion-covid-19-vaccine-janssen_en.pdf Riskienhallintasuunnitelma https://www.ema.europa.eu/en/documents/rmp-summary/comirnaty-epar-risk-management-plan_en.pdf https://www.ema.europa.eu/fi/documents/rmp-summary/covid-19-vaccine-moderna-epar-risk-management-plan_en.pdf https://www.ema.europa.eu/fi/documents/rmp-summary/covid-19-vaccine-astrazeneca-epar-risk-management-plan_en.pdf https://www.ema.europa.eu/documents/rmp-summary/covid-19-vaccine-janssen-epar-risk-management-plan_en.pdf
Tämä artikkeli lähetettiin myös:
https://www.linkedin.com/posts/catherinefrade_%C3%A9clairage-sur-les-donn%C3%A9es-publiques-europ%C3%A9ennes-activity-6783385465841082368-KqWr Blogissani täällä: http://www.catherinefrade.com/blog/2021/04/01/ecclairage-sur-les-donnees-publiques-europeennes-des-amm-conditionnelles-pour-les-4-vaccins-covid -19-31-maaliskuu-2021 / Artiklan 2 mukaan Choletin sairaalan CTIAP on 2. huhtikuuta 2021: http://ctiapchcholet.blogspot.com/2021/04/inedit-exclusif-vaccins-contre-la-covid.html 3 artikla, joka on yhdenmukainen oikeusvaikutuksia koskevan 1 artiklan kanssa: https://www.linkedin.com/posts/catherinefrade_vaccins-covid19-vaccination-activity-6785286370953895936-TqHn Eri verkkosivustojen välittämä 1 artikla: https://www.medias-presse.info/doute-sur-la-qualite-intrinseque-des-produits-presentes-comme-vaccins-anti-covid-et-leurs-procedes-de-fabrication-sur-base- virallisten asiakirjojen julkaisema-Euroopan-lääkeviraston / 141451 / saksaksi: Zweifel an der intrinsischen Qualität von Produkten, die als Anti-Covid-Impfstoffe präsentiert werden | MITTELEUROPAN ALALLA https://www.linkedin.com/posts/chantalattia_ces-vaccins-sont-une-exp%C3%A9rimentation-d%C3%A9lai-activity-6784593281922695168-u10g
Jos tiedät muita artikkelin loppuosia, ilmoita minulle, että voin myös jakaa. Ota yhteyttä saadaksesi lisätietoja virallisten julkisten tietojen tulkinnasta.
Lähde:
https://kapitaali.com/neljan-covid-rokotteen-euroopassa-julkisesti-saatavilla-olevan-datan-tarkastelua-30-3-2021/
0 notes
Text
Kiina ja Venäjä voittamassa rokotediplomatian
COVID-19-rokotteita toivotaan ympäri maailmaapandemian voittamiseksi ja talouden elpymisen aloittamiseksi. Länsimaiset lääkeyhtiöt toimivat itsenäisinä voittoa tavoittelevina yrityksinä, mutta valtiojohtoiset Kiina ja Venäjä ovat valjastaneet rokotteet myös osaksi politiikkaansa. Rokotediplomatian seurauksena Kiinan ja Venäjän taloudellinen ja poliittinen vaikutusvalta on kasvussa erityisesti…

View On WordPress
0 notes
Text
Lääketeollisuuden toimitusjohtaja Sanna Lauslahti: Patentit eivät ole este rokotteiden saatavuudelle tai yhteistyölle
Lääketeollisuuden toimitusjohtaja Sanna Lauslahti: Patentit eivät ole este rokotteiden saatavuudelle tai yhteistyölle
Sanna Lauslahti, toimitusjohtaja, Lääketeollisuus ry Viime aikoina on käyty kiivasta keskustelua siitä, että patentit ovat kääntymässä koronapandemian aikana varsinaista tarkoitustaan vastaan suojella yhteiskuntia. Kirjoituksen mukaan maailman 7,8 miljardin väestön rokottaminen sujuu patenttioikeuksien vuoksi liian hitaasti. Vertauskohteeksi nostetaan HIV-rokotteet, joiden saatavuus kehitysmaissa…

View On WordPress
0 notes