#Id rather die than let my tumblr stop being anonymous and unlinked to me.
Explore tagged Tumblr posts
Visit Tumblr Blog
Explore Tumblr blogs with no restrictions, modern design and the best experience.
Last Seen Tumblr Blogs





Fun Fact
Tumblr has 411 employees.
Text
This is gonna be a long reply, so I'll put a read more in case people don't want to read all this.
Ya this all tracks with what my lab sees (we study intrinsically disordered proteins (IDPs) in the Transcription Factor Co-regulator family along with the Transcription Factors they bind to, but that's less important in the context of this paper). IDPs are just weird proteins. For one, we can actually boil our E. coli to lyse it and separate out whatever protein we’re expressing because there’s so little structure to unfold when boiling, while all the native proteins from the E. coli are (mostly. I’m no an expert on bacterial proteins so maybe some are IDPs) structured and unfold while boiling, exposing their hydrophobic patches and becoming less soluble so they crash out. Then just centrifuge, filter, and run down a HisTrap column and voilà, protein of interest.
HOWEVER! While IDPs don't tend to “aggregate” like structured proteins will, there does seem to be a propensity toward other forms of interaction. There’s still a lot of debate on condensate formation/Liquid-Liquid Phase Separation (that’s a whole nother topic. I'l include a couple references if people want to read about that, just cuz it's a very active field of research and while I have nothing against Wikipedia for quick learning, I don't think Wikipedia is up to date on this) plenty of researchers are convinced IDPs love to form these in vivo to increase local concentration, other groups are finding evidence that IDPs are unique in this ability. BUT my lab has definitely found that IDPs can create gels. We still aren’t sure what causes this, its not just concentration. Might have to do with freezing at too high a concentration? But all that to say that IDPs aren’t necessarily easier to purify: they clog filters like nobody’s business, one of the ones I use a lot seems to crash out if left in a fridge overnight but is stable at room temp(???), despite being mostly polar and charged residues, they hate being labeled with fluorescent molecules and we’ve only had one be happy with a GFP tag (a notoriously easy to use tag that’s a fluorescent protein from jellyfish).
Other quick note before I forget, but idk if they included a linker between the His-Tag and their proteins. This could bias their results toward IDPs, since often you need a linker between the 6xHis and structured proteins (my lab uses a linker that's 10 residues long, but that's to include a cleavage site if we want to remove the His-Tag). Ok, I checked the methods and they only added L and Q, that doesn't seem long enough. The main concern is that if the protein is structured to the very end, it might put the His-Tag in a weird position so it can't bind properly for purification, making the protein seem like it can't be purified. I'm being nitpicky here, but it could have biased their data toward IDPs being "more soluble".
Another thing to remember is that AlphaFold/ESMFold/Insert-Other-Protein-Prediction-Models were trained on the Protein DataBank’s uploaded structures. And the VAST majority of PDB structures are crystal structures, meaning the proteins are essentially “frozen” in place, and then investigated using X-Ray Crystallography (shooting X-Rays at the protein crystals and using the diffraction pattern to solve where all the atoms are). So proteins with lots of tertiary structure form crystals very easily, and since they have a set structure* even in solution, crystallizing them gives lots of replicates of the same shape over and over, like tiles. IDPs however (and intrinsically disordered regions of otherwise structured proteins) don’t make the same repetition since in solution they’re like big long spaghetti noodles**. Once these are “frozen” in the crystals, those floppy bits cane go wherever they can fit, and so you don’t have any confidence on them (if they crystallized in the first place. Hard to form crystals without a repeating solid shape). Since all the protein prediction software is trained on these crystals, there is a MASSIVE bias toward structured proteins and against IDPs and even IDRs.
tl;dr: Protein Prediction Models are designed to show structural information about how protein crystallizes, but not necessarily how it might function in a living system. This isn’t a knock on this blog, its a great blog. Both fun and educational, but there are limits to what AI can do.
*There's still movement on these proteins. No protein is fully frozen, but the small movements found in solution can either (a) be practically non-existent in the crystal or (b) be drastically minimized when adding a ligand, which many proteins need to crystal anyways. Crystallography provides valuable structural information, but it’s static and not necessarily accurate to how the protein behaves in vivo.
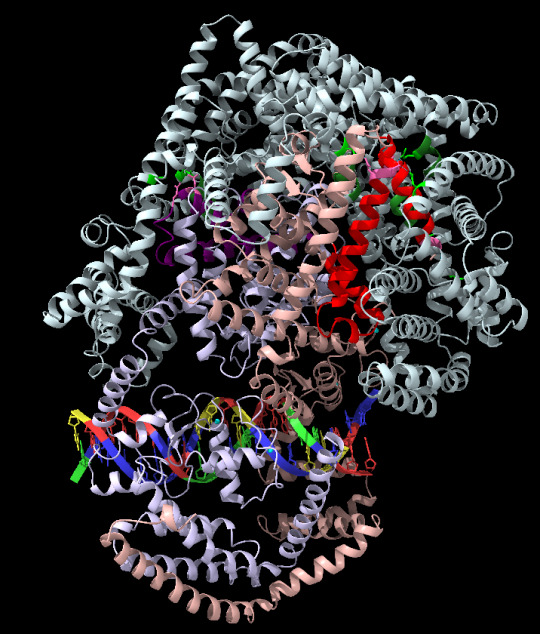
**I actually tested AlphaFold3 when it dropped a couple months ago with the main protein I’m working with and whenever AF3 suggested my protein was condensed down into a bunch of close helices hunting at tertiary structure, I assumed that structure was hallucinated and not accurate. If it’s not a bowl of spaghetti, that’s not my protein. White is my coregulator, peach is it's normal binding partner (the transcription factor), lilac is another TF (these two like to be in a heterodimer), and then there's some DNA at the bottom. Brighter colors were just the regions where binding between the Coreg and TFs occurs. First image is good, second is an AF3 hallucination.

Condensate/LLPS/Etc papers:
Article Review
Okay, I know this isn't what I usually do on here, but I found this amazing article that fits with the theme of this blog so well, and I just had to share and talk about it! it's free to read here:
if you don't want to read the whole thing, i did my best to summarize it here. if you notice any mistakes, please let me know and i'll fix them!
researchers created random protein sequences to study. these were 100 residues long (or 109 with the inclusion of an N-terminal Met and a C-terminal 6His tag) and were made by either sampling different fragments of natural proteins from databases or by combining letters at random. this is not the same as using words, since in this study each letter was chosen independently, and the likelihood of choosing a letter matched the amino acid's relative frequency, but its still a neat comparison to this blog. they elaborate on this more in the methods section for anyone interested!
proteins in their generated library were analyzed using various algorithms to predict the occurrence of alpha helices and beta sheets. they were then sorted by relative disorder and secondary structure content. interestingly, the amount of secondary structure formation was not much lower for random proteins compared to those taken from pieces of databases. the three groups going forward were ordered, disordered, and a random sample.
next, they recombinantly expressed the selected proteins in E. coli and purified them for further analysis. I won't get into the specific assays, but overall they found that the more ordered proteins were more prone to aggregation and oligomerization, while the disordered protein were more likely to be expressed and soluble! following sequence analysis, they also determined that the disordered proteins did tend to deviate from the expected amino acid frequencies, which likely explains their increased level of disorder. because of all this, the less ordered random proteins are likely better suited for future evolution towards some function.
tldr: random proteins can form secondary structures and be expressed in vivo. interestingly, while the more structured newly created proteins were shown to clump together (which is Not Good!) in cells, disordered proteins did not and were actually well tolerated.
given all of that, i think i may have been a bit harsh towards some of the uglier looking structures on here. apparently, we can either have things that look like proteins but cause problems, or we can have ugly messes that are pretty chill for the most part. it still feels incredibly unintuitive to have more trust in the low confidence unstructured sequences, but this new information is still good and interesting to have!
#sorry this reply is so long. i just literally study IDPs so I had to comment#Lol. Firefox doesn't like in vivo but it's ok with e. coli. sure why not#Sorry I'm not adding the names of my proteins. I dont want this to pop up if my labmates are searching for new research on our proteins.#if any coworker ever finds this. 1) don't ever tell me. Act like you didnt. 2) just kill me. Please.#Id rather die than let my tumblr stop being anonymous and unlinked to me.
127 notes
·
View notes