#Enzyme Products Database
Explore tagged Tumblr posts
Visit Tumblr Blog
Explore Tumblr blogs with no restrictions, modern design and the best experience.
Last Seen Tumblr Blogs





Fun Fact
US Tumblr user growth rate is estimated to slow down to 4.1%.
Text
Why Understanding API Consumption is Crucial for R&D and Regulatory Strategy
API consumption data is more than an operational metric. It’s a strategic tool for innovation and compliance in today’s pharma landscape.
Understanding which APIs dominate the market—why they do, where they’re used most, and how they’re evolving—is crucial for any organization serious about optimizing its R&D pipeline and regulatory positioning.By aligning science with real-world usage and regulation with market needs, pharma leaders can unlock smarter, faster, and more resilient drug development pathways.
#Inorganic Chemicals Database#Food-Additives Database#Fertilizers Database#Excipients Database#Enzyme Products Database
0 notes
Text
IdentificationPhysical DataSpectraRoute of Synthesis (ROS)Safety and HazardsOther Data Identification Product NameBovin beta-1,4-Galactosyltransferase 1 (Y289L) Molecular StructureCAS Registry Number 24-1-1060SynonymsMolecular FormulaMolecular WeightInChIInChI KeyCanonical SMILES Physical Data AppearanceWhite freeze-dried powder Spectra No data available Route of Synthesis (ROS) No data available Safety and Hazards GHS Hazard StatementsNot Classified Source: European Chemicals Agency (ECHA)License Note: Use of the information, documents and data from the ECHA website is subject to the terms and conditions of this Legal Notice, and subject to other binding limitations provided for under applicable law, the information, documents and data made available on the ECHA website may be reproduced, distributed and/or used, totally or in part, for non-commercial purposes provided that ECHA is acknowledged as the source: “Source: European Chemicals Agency, http://echa.europa.eu/”. Such acknowledgement must be included in each copy of the material. ECHA permits and encourages organisations and individuals to create links to the ECHA website under the following cumulative conditions: Links can only be made to webpages that provide a link to the Legal Notice page.License URL: https://echa.europa.eu/web/guest/legal-noticeRecord Name: (1-Cyano-2-ethoxy-2-oxoethylidenaminooxy)dimethylamino-morpholino-carbenium hexafluorophosphateURL: https://echa.europa.eu/information-on-chemicals/cl-inventory-database/-/discli/details/213446Description: The information provided here is aggregated from the “Notified classification and labelling” from ECHA’s C&L Inventory. Read more: https://echa.europa.eu/information-on-chemicals/cl-inventory-database Other Data Use PatternBovine B4GALT1(Y289L) is a mutant galactosyltransferase capable of efficiently transferring GalNAc or GalNAz onto terminal GlcNAc residues. This enzyme is particularly useful in the synthesis of antibody-drug conjugates (ADCs), which are applied in targeted cancer therapies and other biomedical applications. Read the full article
0 notes
Text
The Clean Sweep: A Report on the Japan Medical Device Cleaning Market
Japan Medical Device Cleaning Market Growth & Trends
The Japan Medical Device Cleaning Market size is anticipated to reach USD 1,169.49 million by 2030 and is projected to grow at a CAGR of 10.83% over the forecast period, according to a new report by Grand View Research, Inc. This growth can be attributed to the increasing competition among the market players and the growing efforts to reduce hospital-acquired infections.
Several studies are being published focusing on the increasing number of infections acquired in hospitals and the measures that can prevent them. For instance, a study published by the National Library of Medicine in May 2020 found that nosocomial infections, also known as healthcare-acquired infections, are a significant burden on hospitalized patients in Japan. This study analyzed the Japanese claims database and found that out of 73,962,409 inpatients registered in the database, 9.7% had community-acquired infections (CAI), and 4.7% had nosocomial infections (NI). As a result, the growing burden of hospital-acquired infections is expected to increase the demand for medical device cleaning products in Japan.
Moreover, the growing use of single-use devices and the rising manufacturing of medical devices are anticipated to propel the Japanese medical device cleaning market. Industry stakeholders are focusing on increasing the development of medical devices in the country. For instance, in May 2023, Terumo Corporation, a medical device company, invested around USD 360 million to construct a new manufacturing facility for the Medical Care Solutions Company in Japan.
In addition, in January 2022, Kaneka Corporation invested around USD 69 million to build a new medical device plant in the Tomatoh Industrial Area in the northern area of Japan. Companies are expanding their manufacturing facilities. These expanding manufacturing facilities will require medical device cleaning solutions for sterilization, reprocessing, and cleaning in the future. Thus, the rise in investments in medical device manufacturing and development in Japan is projected to increase the demand for medical device cleaning products in the coming years.
Curious about the Japan Medical Device Cleaning Market? Download your FREE sample copy now and get a sneak peek into the latest insights and trends.
Japan Medical Device Cleaning Market Report Highlights
Based on device type, the semi-critical segment dominated the market in 2023 and accounted for 46.02% of revenue share. However, the critical segment is anticipated to grow fastest over the forecast period due to the increasing infection control awareness and growing aging population.
Based on technique, the disinfection segment dominated the market in 2023 and accounted for 49.53% of the revenue share. However, the sterilization segment is anticipated to grow fastest from 2024 to 2030. Advancements in sterilization technologies are expected to boost segment growth in the coming years.
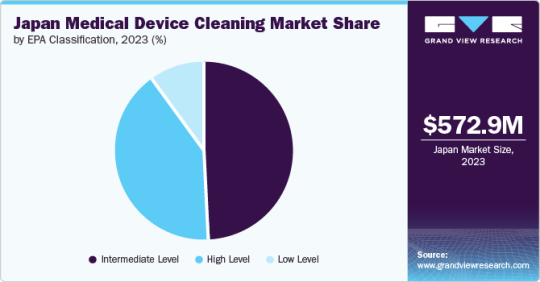
Based on EPA classification, the intermediate-level segment dominated the Japan market and accounted for the largest revenue share, 48.84%, in 2023. In contrast, the high-level segment is expected to grow fastest, with the fastest CAGR over the forecast period.
Japan Medical Device Cleaning Market Segmentation
Grand View Research has segmented the Japan Medical device cleaning market based on the device type, technique, and EPA classification:
Japan Medical Device Cleaning Device Type Outlook (Revenue, USD Million, 2018 - 2030)
Non-Critical
Semi-Critical
Critical
Japan Medical Device Cleaning Technique Outlook (Revenue, USD Million, 2018 - 2030)
Cleaning
Detergents
Buffers
Chelators
Enzymes
Others
Disinfection
Chemical
Alcohol
Chlorine & Chorine Compounds
Aldehydes
Others
Metal
Ultraviolet
Others
Sterilization
Heat Sterilization
Ethylene Dioxide (ETO) Sterilization
Radiation Sterilization
Japan Medical Device Cleaning EPA Classification Outlook (Revenue, USD Million, 2018 - 2030)
High Level
Intermediate Level
Low Level
Download your FREE sample PDF copy of the Japan Medical Device Cleaning Market today and explore key data and trends.
0 notes
Text
Eco-Friendly Alkaline Protease: Production and Application in Detergents

Abstract
An alkalophilic, halotolerant bacterial strain ASM1 isolated from agricultural soil was found to be capable of producing extracellular protease enzyme. Proteolytic strain was identified as Bacillus cereus and nucleotide sequence has been submitted in NCBI database under accession number KJ600795. Optimum enzyme production in terms of specific activity 9.58 U/mg of total protein was obtained at 35°C; pH, 9.0; 1 % glucose as C-source and 35 g/l beef extract as N-source after 48 hours of incubation in a defined medium inoculated with 2% inoculum size. Bacterial isolate was capable of tolerating up to 12.5% NaCl without requiring salt for physiological activities. Bacterial crude enzyme was purified by 6 folds with 25% yield and specific activity of 57.9 U/mg protein by two step purification i.e. ammonium sulfate precipitation and gel-filtration chromatography. Thermostability studies revealed retention of 60% proteolytic activity upto 55°C. Moreover enzyme remained stable in the pH range of 6-11. PMSF (phenylmethylsulfonyl fluoride) inhibited enzyme activity categorizing the enzyme as a serine protease. Enzyme remained stable in presence of 8 different metals, however activity declined in the presence of 20 mM Fe2+ ions. Enzyme retained substantial stability in the presence of solvents, surfactants, commercially available detergents, and NaCl. Enzyme exhibited efficacious de-staining of fixed blood stains in the washing test at room temperature, without requiring additional energy. This particular type of protease enzyme is of immense importance due to its alkaline-halotolerant profile at mesophilic temperature range which is a great deal for revolutionizing detergents’ industry.

Introduction
Proteases are hydrolytic enzymes that catalyze hydrolysis of proteins by addition of water across the peptide bonds into smaller polypeptides and free amino acids (Beg and Gupta, 2003). Proteases are ubiquitous in nature playing important physiological roles, in all domains of life (Barrett et al., 2001; Burhan et al., 2003). Microbial proteases constitute one of the three commercially significant groups of enzymes, contributing more than 60% of share in the global enzyme market (Chu, 2007; Huang et al., 2003; Jayakumar et al., 2012; Jon, 2008). Proteases constitute a very diverse group of biocatalysts with members having different substrate specificities; nature of catalytic sites; evolutionary relationship in amino acids’ sequence; catalytic mechanisms and varying activity-stability profiles on broad range of temperature and pH (Rai and Mukherjee, 2010; Rao and Narasu, 2007; Rawlings et al., 2012).
Bacterial bio-factories hold much more temptation for exploitation than other enzyme producers due to the ease of handling and production in a limited time and space with less complicated purification steps. Besides that bacteria are susceptible to artificial genetic manipulations and are able to survive under diverse and extreme environmental conditions (Burhan et al., 2003; Khademi et al., 2013; Rao and Narasu, 2007; Rao et al., 1998). Genus Bacillus is considered as the most significant source of bulk amounts of industrially important neutral and alkaline proteases which are highly stable at temperature and pH extremes (Beg and Gupta, 2003; Gupta and Khare, 2007; Venugopal and Saramma, 2006; Yang et al., 2000).

Proteases active and stable in the alkaline pH range are referred as alkaline proteases. Active site of alkaline protesaes may contain serine residues or metal ions (Khan, 2013). Alkaline proteases with serine residues on catalytic site are referred as Serine Alkaline Proteases (SAPs). Optimum pH for production and activity of serine proteases ranges between pH 7.0-12.0. Some SAPs are endowed with additional characteristic of halotolerance which makes them perfect tool for utilization in various industrial processes (Joo and Chang, 2005; Joshi et al., 2007; Maurer, 2004; Purohit and Singh, 2011; Singh et al., 2010). Stability studies in presence of salts, metal ions, surfactants, oxidants and solvents help in prospecting probable use of enzyme in industry (Gupta and Khare, 2007; Joo et al., 2003; Zambare et al., 2014). Alkaline proteases are majorly used as additives in the commercial detergents (Maurer, 2004). Different industries especially leather and detergent industries require efficacious, environment friendly and economical approaches for degradation of unwanted proteins (Hameed et al., 1996; Huang et al., 2003; Wang et al., 2007).
Protease production can be enhanced by optimization and manipulation of fermentation methods and conditions; cloning and modulation of genes expression and protein engineering (Gupta et al., 2002a; Gupta et al., 2002b). To achieve high protease production rates, understanding of strategies for protease production and broad range application in the industrial processes hold central importance. Aim of this study was to isolate, characterize and optimize proteolytic strain present in soil biome for enhanced enzyme production. Moreover, biochemical characterization and stability studies of the enzyme were aimed to determine possible eco-friendly application of enzyme in detergent industry.
Source : Eco-Friendly Alkaline Protease: Production and Application in Detergents | InformatiiveBD
1 note
·
View note
Text
Transforming Cardiac Diagnostics: The Rise of Point-of-Care Testing.
Cardiac POC is called as Cardiac Point of care which used to diagnose patients. POC includes cholesterol and coagulation testing as well as brain Natriuretic peptide test and troponin. Cardiac Point of care help in detection of enzyme detection which is relapsed by during cardiac condition like lactate, and troponin.
Request Sample link:
https://qualiketresearch.com/request-sample/POC-Cardiac-Market/request-sample
Key Players:
Abbott Laboratories, Medtronic Inc, Roche Diagnostics, Boston Scientific Corp., Alere Inc, and Others
Top Regional scope:
NorthAmerica
Asia Pacific
Europe
Latin America
Middle East
By Drivers Global POC Cardiac Market:
Change in healthcare regulatory standards across the world and practitioner approach towards POC diagnostics is the key driving factor which is expected to boost the global POC cardiac market growth. Furthermore, Rise in government investments on development of healthcare infrastructure in order to facilitate advanced facilities will have the positive impact on market growth. Moreover, increase in incidences of chronic diseases will fuel the market growth. In addition to that, continuous technological advancements by key players expected to propel market growth during this forecast period.
By Restraints Global POC Cardiac Market:
However, lack of skilled professionals is the major challenging factor which is expected to hamper the global POC cardiac market. Also, lack of healthcare infrastructure in developing countries will affect the market growth.
Read More Info Link:
https://qualiketresearch.com/reports-details/POC-Cardiac-Market
By Product:
Cardiac Markers Test
Combinational Test Kits
Myoglobin Test
Brain Natriuretic Peptide (BNP) Test
Creatine Kinase MB isoenzyme (CK-MB) Test
Cardiac troponin Test
Analyzers
By Application:
Hospitals
Research Laboratories
Ambulatory Surgical Centers
Diagnostic Laboratories
Client Attention:
What is market research?
Gathering, evaluating, and interpreting data on a market, a product or service that will be sold in that market, and the past, present, and potential clients for the product or service constitute the process of conducting market research.
What are the different types of market research?
Primary research, secondary research, qualitative research, and quantitative research are some of the several kinds of market research.
What is secondary research?
Secondary research involves gathering information from already published sources, such online databases, industry journals, and market reports.
About Us:
QualiKet Research is a leading Market Research and Competitive Intelligence partner helping leaders across the world to develop robust strategy and stay ahead for evolution by providing actionable insights about ever changing market scenario, competition and customers.
QualiKet Research is dedicated to enhancing the ability of faster decision making by providing timely and scalable intelligence.
QualiKet Research strive hard to simplify strategic decisions enabling you to make right choice. We use different intelligence tools to come up with evidence that showcases the threats and opportunities which helps our clients outperform their competition. Our experts provide deep insights which is not available publicly that enables you to take bold steps.
Contact Us:
6060 N Central Expy #500 TX 75204, U.S.A
+1 214 660 5449
1201, City Avenue, Shankar Kalat Nagar,
Wakad, Pune 411057, Maharashtra, India
+91 9284752585
Sharjah Media City , Al Messaned, Sharjah, UAE.
+971 568464312
0 notes
Text
Hydroquinone Prices | Pricing | Trend | News | Database | Chart | Forecast
Hydroquinone is a chemical compound widely used in skincare products for its skin-lightening properties. It is primarily used to treat conditions such as hyperpigmentation, melasma, freckles, and age spots. Hydroquinone works by inhibiting the enzyme tyrosinase, which is responsible for the production of melanin, the pigment that gives skin its color. As the demand for hydroquinone has increased in recent years, especially with rising awareness of skincare routines, the price of hydroquinone has become a subject of interest for many, particularly in the cosmetics and pharmaceutical industries. The prices of hydroquinone can fluctuate due to a variety of factors, including raw material costs, manufacturing processes, regulatory policies, supply and demand dynamics, and geographical influences.
Hydroquinone prices are largely influenced by the availability and cost of raw materials. Hydroquinone is synthesized through various chemical processes, and the availability of key chemicals, such as benzene or phenol, directly impacts production costs. When these raw materials experience price hikes due to supply shortages, geopolitical issues, or changes in environmental regulations, the price of hydroquinone rises correspondingly. This chain reaction from raw material costs to the final product pricing makes it important for manufacturers to secure stable sources of these inputs.
Get Real Time Prices for Hydroquinone : https://www.chemanalyst.com/Pricing-data/hydroquinone-1392
Another factor that influences hydroquinone pricing is the scale and efficiency of the manufacturing processes. Hydroquinone production involves complex chemical reactions, and companies with more advanced technologies can often produce the compound at a lower cost. This competitive advantage allows larger manufacturers to offer more competitive pricing while smaller or less technologically advanced producers may struggle to keep their prices low. Furthermore, fluctuations in energy costs also impact production expenses, as hydroquinone synthesis often requires high levels of energy for processing. If energy prices spike, manufacturers may increase product prices to compensate for the added expense.
Regulatory policies also play a critical role in determining hydroquinone prices. In many countries, hydroquinone is tightly regulated due to concerns about its safety when used in high concentrations or over extended periods. Some regions, such as the European Union, have banned the sale of over-the-counter products containing hydroquinone due to potential health risks, while others have imposed strict limits on allowable concentrations. In markets where hydroquinone is highly regulated, compliance costs for manufacturers, including quality control measures, safety testing, and certifications, can drive up production expenses. Consequently, companies operating in these regions may charge more for their products compared to those in less regulated markets.
On the demand side, the popularity of hydroquinone in skincare products is a major driver of price trends. As consumers become more conscious of skincare ingredients and seek solutions for skin conditions like dark spots and uneven skin tone, the demand for products containing hydroquinone has risen. This increase in demand has created a market where prices can rise, especially if supply struggles to keep pace. For cosmetic companies, this heightened demand can lead to higher wholesale prices for hydroquinone, which are then passed on to consumers through more expensive skincare products.
Geographical differences also contribute to variations in hydroquinone prices. Countries with robust chemical manufacturing sectors, such as China and India, often have lower production costs due to economies of scale and access to cheaper raw materials. As a result, hydroquinone sourced from these regions may be more affordable. Conversely, in countries with stricter environmental regulations, higher labor costs, or limited access to raw materials, the price of hydroquinone can be significantly higher. Additionally, shipping and logistics costs, as well as import taxes, can further contribute to the price disparities between different regions.
The impact of market competition also cannot be ignored when analyzing hydroquinone prices. As with many commodities, competition among manufacturers and suppliers can help to stabilize prices or, in some cases, drive them down. When several manufacturers compete for market share, they may offer lower prices to attract buyers. However, in markets where competition is limited or where a few key players dominate, prices can be kept artificially high. In such scenarios, consumers and businesses may face higher costs for hydroquinone-based products.
One more factor affecting hydroquinone prices is the increasing trend toward natural or alternative skin-lightening ingredients. With growing awareness of the potential side effects of hydroquinone, some consumers are shifting toward products that contain natural alternatives like kojic acid, arbutin, or licorice extract. This trend could reduce the demand for hydroquinone in certain markets, potentially leading to lower prices as manufacturers adjust to decreased demand. However, in markets where hydroquinone remains the most effective and popular skin-lightening ingredient, prices may continue to remain high.
In conclusion, the price of hydroquinone is influenced by a complex interplay of factors, including raw material costs, manufacturing efficiency, regulatory policies, supply and demand dynamics, and geographical considerations. The growing demand for hydroquinone in skincare, coupled with competition in the marketplace, will likely continue to shape its pricing trends in the foreseeable future. With the rise of alternative skin-lightening ingredients, it remains to be seen whether hydroquinone prices will stabilize, decrease, or experience further fluctuations. However, for now, hydroquinone remains a key ingredient in many skin treatments, and its price is a reflection of the global forces at play in the beauty and pharmaceutical industries.
Get Real Time Prices for Hydroquinone : https://www.chemanalyst.com/Pricing-data/hydroquinone-1392
Contact Us:
ChemAnalyst
GmbH - S-01, 2.floor, Subbelrather Straße,
15a Cologne, 50823, Germany
Call: +49-221-6505-8833
Email: [email protected]
Website: https://www.chemanalyst.com
#Hydroquinone#Hydroquinone Price#Hydroquinone Prices#Hydroquinone Price Monitor#Hydroquinone News#Hydroquinone Database#Hydroquinone Price Chart
0 notes
Text
Characterization of a marine bacteria through a novel metabologenomics approach
Exploiting microbial natural products is a key pursuit of the bioactive compound discovery field. Recent advances in modern analytical techniques have increased the volume of microbial genomes and their encoded biosynthetic products measured by mass spectrometry-based metabolomics. However, connecting multi-omics data to uncover metabolic processes of interest is still challenging. This results in a large portion of genes and metabolites remaining unannotated. Further exacerbating the annotation challenge, databases and tools for annotation and omics integration are scattered, requiring complex computations to annotate and integrate omics datasets. Here we performed a two-way integrative analysis combining genomics and metabolomics data to describe a new approach to characterize the marine bacterial isolate BRA006 and to explore its biosynthetic gene cluster (BGC) content as well as the bioactive compounds detected by metabolomics. We described BRA006 genomic content and structure by comparing Illumina and Oxford Nanopore MinION sequencing approaches. Digital DNA:DNA hybridization (dDDH) taxonomically assigned BRA006 as a potential new species of the Micromonospora genus. Starting from LC-ESI(+)-HRMS/MS data, and mapping the annotated enzymes and metabolites belonging to the same pathways, our integrative analysis allowed us to correlate the compound Brevianamide F to a new BGC, previously assigned to other function. http://dlvr.it/TBpG2S
0 notes
Text
Medicated Feed Additives Market Size, Share, Comprehensive Analysis, Opportunity Assessment By 2030

The market research study titled “Medicated Feed Additives Market Share, Trends, and Outlook | 2031,” guides organizations on market economics by identifying current Medicated Feed Additives market size, total market share, and revenue potential. This further includes projections on future market size and share in the estimated period. The company needs to comprehend its clientele and the demand it creates to focus on a smaller selection of items. Through this chapter, market size assists businesses in estimating demand in specific marketplaces and comprehending projected patterns for the future.
The Medicated Feed Additives market report also provides in-depth insights into major industry players and their strategies because we understand how important it is to remain ahead of the curve. Companies may utilize the objective insights provided by this market research to identify their strengths and limitations. Companies that can capitalize on the fresh perspective gained from competition analysis are more likely to have an edge in moving forward.
With this comprehensive research roadmap, entrepreneurs and stakeholders can make informed decisions and venture into a successful business. This research further reveals strategies to help companies grow in the Medicated Feed Additives market.
Market Analysis and Forecast
This chapter evaluates several factors that impact on business. The economics of scale described based on market size, growth rate, and CAGR are coupled with future projections of the Medicated Feed Additives market. This chapter is further essential to analyze drivers of demand and restraints ahead of market participants. Understanding Medicated Feed Additives market trends helps companies to manage their products and position themselves in the market gap.
This section offers business environment analysis based on different models. Streamlining revenues and success is crucial for businesses to remain competitive in the Medicated Feed Additives market. Companies can revise their unique selling points and map the economic, environmental, and regulatory aspects.
Report Attributes
Details
Segmental Coverage
Mixture Type
Supplements
Concentrates
Premix Feeds
Base Mixes
Type
Antioxidants
Antibiotics
Probiotics and Prebiotics
Enzymes
Amino Acids
Others
Category
Category I
Category II
Livestock
Ruminants
Poultry
Swine
Aquaculture
Others
Geography
North America
Europe
Asia Pacific
and South and Central America
Regional and Country Coverage
North America (US, Canada, Mexico)
Europe (UK, Germany, France, Russia, Italy, Rest of Europe)
Asia Pacific (China, India, Japan, Australia, Rest of APAC)
South / South & Central America (Brazil, Argentina, Rest of South/South & Central America)
Middle East & Africa (South Africa, Saudi Arabia, UAE, Rest of MEA)
Market Leaders and Key Company Profiles
Adisseo France Sas
Alltech Inc. (Ridley)
Archer Daniels Midland Company
Biostadt India Limited
Cargill, Incorporated
CHS Inc.
Hipro Animal Nutrition
Purina Animal Nutrition (Land O' Lakes)
Zagro
Zoetis Inc.
Other key companies
Our Unique Research Methods at The Insight Partners
We offer syndicated market research solutions and consultation services that provide complete coverage of global markets. This report includes a snapshot of global and regional insights. We pay attention to business growth and partner preferences, that why we offer customization on all our reports to meet individual scope and regional requirements.
Our team of researchers utilizes exhaustive primary research and secondary methods to gather precise and reliable information. Our analysts cross-verify facts to ensure validity. We are committed to offering actionable insights based on our vast research databases.
Strategic Recommendations
Strategic planning is crucial for business success. This section offers strategic recommendations needed for businesses and investors. Forward forward-focused vision of a business is what makes it through thick and thin. Knowing business environment factors helps companies in making strategic moves at the right time in the right direction.
Summary:
Medicated Feed Additives Market Forecast and Growth by Revenue | 2031
Market Dynamics – Leading trends, growth drivers, restraints, and investment opportunities
Market Segmentation – A detailed analysis by product, types, end-user, applications, segments, and geography
Competitive Landscape – Top key players and other prominent vendors
About Us:
The Insight Partners is a one-stop industry research provider of actionable intelligence. We help our clients in getting solutions to their research requirements through our syndicated and consulting research services. We specialize in industries such as Semiconductor and Electronics, Aerospace and Defense, Automotive and Transportation, Biotechnology, Healthcare IT, Manufacturing and Construction, Medical Devices, Technology, Media and Telecommunications, Chemicals and Materials.
Contact Us: www.theinsightpartners.com
0 notes
Text
Unlock the Secrets of Pharmaceutical Marketing with Chemxpert Database
Dive into the world of pharmaceutical marketing with Chemxpert Database. Discover insights on the biggest pharmaceutical companies and the top 10 pharmaceutical companies globally. Stay updated on pharma marketing trends and explore detailed profiles of pharmaceutical companies in Germany. With Chemxpert Database, gain a competitive edge in the ever-evolving pharmaceutical industry.
#Industrial Chemicals Database#Food-Additives Database#Excipients Database#Enzyme Products Database#Dyes Chemical Database
1 note
·
View note
Text
“Rosetta Stone” of cell signaling could expedite precision cancer medicine
New Post has been published on https://sunalei.org/news/rosetta-stone-of-cell-signaling-could-expedite-precision-cancer-medicine/
“Rosetta Stone” of cell signaling could expedite precision cancer medicine

A newly complete database of human protein kinases and their preferred binding sites provides a powerful new platform to investigate cell signaling pathways.
Culminating 25 years of research, MIT, Harvard University, and Yale University scientists and collaborators have unveiled a comprehensive atlas of human tyrosine kinases — enzymes that regulate a wide variety of cellular activities — and their binding sites.
The addition of tyrosine kinases to a previously published dataset from the same group now completes a free, publicly available atlas of all human kinases and their specific binding sites on proteins, which together orchestrate fundamental cell processes such as growth, cell division, and metabolism.
Now, researchers can use data from mass spectrometry, a common laboratory technique, to identify the kinases involved in normal and dysregulated cell signaling in human tissue, such as during inflammation or cancer progression.
“I am most excited about being able to apply this to individual patients’ tumors and learn about the signaling states of cancer and heterogeneity of that signaling,” says Michael Yaffe, who is the David H. Koch Professor of Science at MIT, the director of the MIT Center for Precision Cancer Medicine, a member of MIT’s Koch Institute for Integrative Cancer Research, and a senior author of the new study. “This could reveal new druggable targets or novel combination therapies.”
The study, published in Nature, is the product of a long-standing collaboration with senior authors Lewis Cantley at Harvard Medical School and Dana-Farber Cancer Institute, Benjamin Turk at Yale School of Medicine, and Jared Johnson at Weill Cornell Medical College.
The paper’s lead authors are Tomer Yaron-Barir at Columbia University Irving Medical Center, and MIT’s Brian Joughin, with contributions from Kontstantin Krismer, Mina Takegami, and Pau Creixell.
Kinase kingdom
Human cells are governed by a network of diverse protein kinases that alter the properties of other proteins by adding or removing chemical compounds called phosphate groups. Phosphate groups are small but powerful: When attached to proteins, they can turn proteins on or off, or even dramatically change their function. Identifying which of the almost 400 human kinases phosphorylate a specific protein at a particular site on the protein was traditionally a lengthy, laborious process.
Beginning in the mid 1990s, the Cantley laboratory developed a method using a library of small peptides to identify the optimal amino acid sequence — called a motif, similar to a scannable barcode — that a kinase targets on its substrate proteins for the addition of a phosphate group. Over the ensuing years, Yaffe, Turk, and Johnson, all of whom spent time as postdocs in the Cantley lab, made seminal advancements in the technique, increasing its throughput, accuracy, and utility.
Johnson led a massive experimental effort exposing batches of kinases to these peptide libraries and observed which kinases phosphorylated which subsets of peptides. In a corresponding Nature paper published in January 2023, the team mapped more than 300 serine/threonine kinases, the other main type of protein kinase, to their motifs. In the current paper, they complete the human “kinome” by successfully mapping 93 tyrosine kinases to their corresponding motifs.
Next, by creating and using advanced computational tools, Yaron-Barir, Krismer, Joughin, Takegami, and Yaffe tested whether the results were predictive of real proteins, and whether the results might reveal unknown signaling events in normal and cancer cells. By analyzing phosphoproteomic data from mass spectrometry to reveal phosphorylation patterns in cells, their atlas accurately predicted tyrosine kinase activity in previously studied cell signaling pathways.
For example, using recently published phosphoproteomic data of human lung cancer cells treated with two targeted drugs, the atlas identified that treatment with erlotinib, a known inhibitor of the protein EGFR, downregulated sites matching a motif for EGFR. Treatment with afatinib, a known HER2 inhibitor, downregulated sites matching the HER2 motif. Unexpectedly, afatinib treatment also upregulated the motif for the tyrosine kinase MET, a finding that helps explain patient data linking MET activity to afatinib drug resistance.
Actionable results
There are two key ways researchers can use the new atlas. First, for a protein of interest that is being phosphorylated, the atlas can be used to narrow down hundreds of kinases to a short list of candidates likely to be involved. “The predictions that come from using this will still need to be validated experimentally, but it’s a huge step forward in making clear predictions that can be tested,” says Yaffe.
Second, the atlas makes phosphoproteomic data more useful and actionable. In the past, researchers might gather phosphoproteomic data from a tissue sample, but it was difficult to know what that data was saying or how to best use it to guide next steps in research. Now, that data can be used to predict which kinases are upregulated or downregulated and therefore which cellular signaling pathways are active or not.
“We now have a new tool now to interpret those large datasets, a Rosetta Stone for phosphoproteomics,” says Yaffe. “It is going to be particularly helpful for turning this type of disease data into actionable items.”
In the context of cancer, phosophoproteomic data from a patient’s tumor biopsy could be used to help doctors quickly identify which kinases and cell signaling pathways are involved in cancer expansion or drug resistance, then use that knowledge to target those pathways with appropriate drug therapy or combination therapy.
Yaffe’s lab and their colleagues at the National Institutes of Health are now using the atlas to seek out new insights into difficult cancers, including appendiceal cancer and neuroendocrine tumors. While many cancers have been shown to have a strong genetic component, such as the genes BRCA1 and BRCA2 in breast cancer, other cancers are not associated with any known genetic cause. “We’re using this atlas to interrogate these tumors that don’t seem to have a clear genetic driver to see if we can identify kinases that are driving cancer progression,” he says.
Biological insights
In addition to completing the human kinase atlas, the team made two biological discoveries in their recent study. First, they identified three main classes of phosphorylation motifs, or barcodes, for tyrosine kinases. The first class is motifs that map to multiple kinases, suggesting that numerous signaling pathways converge to phosphorylate a protein boasting that motif. The second class is motifs with a one-to-one match between motif and kinase, in which only a specific kinase will activate a protein with that motif. This came as a partial surprise, as tyrosine kinases have been thought to have minimal specificity by some in the field.
The final class includes motifs for which there is no clear match to one of the 78 classical tyrosine kinases. This class includes motifs that match to 15 atypical tyrosine kinases known to also phosphorylate serine or threonine residues. “This means that there’s a subset of kinases that we didn’t recognize that are actually playing an important role,” says Yaffe. It also indicates there may be other mechanisms besides motifs alone that affect how a kinase interacts with a protein.
The team also discovered that tyrosine kinase motifs are tightly conserved between humans and the worm species C. elegans, despite the species being separated by more than 600 million years of evolution. In other words, a worm kinase and its human homologue are phosphorylating essentially the same motif. That sequence preservation suggests that tyrosine kinases are highly critical to signaling pathways in all multicellular organisms, and any small change would be harmful to an organism.
The research was funded by the Charles and Marjorie Holloway Foundation, the MIT Center for Precision Cancer Medicine, the Koch Institute Frontier Research Program via L. Scott Ritterbush, the Leukemia and Lymphoma Society, the National Institutes of Health, Cancer Research UK, the Brain Tumour Charity, and the Koch Institute Support (core) grant from the National Cancer Institute.
0 notes
Text
The Role of Machine Learning in Catalysis: Advancing Discovery and Optimization

Harnessing the Power of Artificial Intelligence to Transform Catalysis
Advancements in catalysis research have the potential to revolutionize various industries, from energy production to pharmaceuticals. Catalysis plays a crucial role in accelerating chemical reactions, improving efficiency, and reducing environmental impact. Traditionally, catalysis research has relied on empirical knowledge and trial-and-error approaches.
However, with the advent of machine learning and artificial intelligence (AI), there has been a paradigm shift in how catalysis is studied and optimized. Machine learning algorithms have the ability to analyze vast amounts of data, identify patterns, and make predictions, leading to the discovery of new catalysts and the optimization of existing ones. In this article, we will explore the applications of machine learning in catalysis and how it is transforming the field.
youtube
Predictive Modeling and Catalyst Design
One of the key areas where machine learning has made significant contributions is in predictive modeling and catalyst design. By training algorithms on large datasets of experimental and computational data, researchers can develop models that can accurately predict the performance of catalysts under different conditions. These models take into account various factors such as catalyst composition, structure, and reaction conditions to provide insights into catalytic activity and selectivity.
This approach allows researchers to screen a vast chemical space and identify promising catalyst candidates for specific reactions.
For example, a study by Ahneman et al. used machine learning to predict reaction performance in C-N cross-coupling reactions. By training the algorithm on a dataset of experimental results, the researchers were able to identify key features that contribute to high reaction yields.
This approach not only accelerated the discovery of high-performing catalysts but also provided insights into the underlying reaction mechanisms.
Another study by Kim et al. employed active learning combined with experiments to search for an optimal multi-metallic alloy catalyst. The algorithm iteratively selected new catalyst compositions to synthesize and tested their performance, using the feedback to improve its predictions.
This approach allowed the researchers to rapidly explore a large chemical space and identify a highly efficient catalyst for a specific reaction.
Accelerating Materials Discovery
Machine learning has also played a crucial role in accelerating materials discovery in catalysis. By analyzing large databases of materials properties and performance, algorithms can identify trends and correlations that can guide the design of new materials with desired catalytic properties. This approach has been particularly valuable in the field of heterogeneous catalysis, where the catalyst is in a different phase from the reactants.
For example, the Open Catalyst 2020 (OC20) dataset, developed by Chanussot et al., consists of nearly 1.3 million density functional theory (DFT) relaxations of materials surfaces and adsorbates. This extensive database allows researchers to explore a wide range of materials and reactions, providing valuable insights into the factors that influence catalytic activity and selectivity.
Furthermore, machine learning has been used to analyze X-ray absorption spectra and transmission electron microscopy images of catalysts. These techniques enable researchers to gain a deeper understanding of catalyst structures and their evolution during reactions. Mitchell et al.
developed an automated image analysis method for single-atom detection in catalytic materials using transmission electron microscopy. This approach allows for the identification and characterization of catalytic active sites at the atomic scale, providing valuable insights for catalyst design and optimization.
Enzyme Engineering and Biocatalysis
Machine learning has also been applied to enzyme engineering and biocatalysis, offering new possibilities for the design of highly efficient biocatalysts. By training algorithms on large datasets of enzyme sequences and structures, researchers can develop models that can predict enzyme properties and guide the engineering of novel enzymes with desired functionalities.
For example, Wu et al. used machine learning-assisted directed protein evolution with combinatorial libraries to engineer enzymes for improved properties. The algorithm guided the design of protein libraries and selected variants with desired characteristics for further experimental testing.
This approach allowed for the rapid optimization of enzymes for specific reactions, opening up new possibilities for the synthesis of valuable compounds.
Similarly, Lu et al. employed machine learning-aided engineering to design hydrolases for the depolymerization of polyethylene terephthalate (PET). By training the algorithm on a large dataset of enzyme sequences and PET degradation activities, the researchers were able to predict enzyme variants with enhanced PET-degrading capabilities.
This approach offers a promising solution for the recycling of PET, a major environmental challenge.
Challenges and Opportunities
While machine learning has shown great promise in catalysis research, there are still challenges that need to be addressed. One of the main challenges is the availability and quality of data. High-quality datasets are essential for training accurate machine learning models, but obtaining such datasets can be time-consuming and expensive.
Furthermore, there is a need for standardized data formats and protocols to enable data sharing and collaboration among researchers.
Another challenge is the interpretability of machine learning models. While these models can make accurate predictions, understanding the underlying factors that contribute to the predictions can be challenging. Interpretable machine learning methods, such as decision trees and rule-based models, can help address this issue by providing explanations for the model's predictions.
Despite these challenges, the opportunities offered by machine learning in catalysis research are immense. By combining experimental data, computational modeling, and machine learning algorithms, researchers can accelerate the discovery of new catalysts, optimize existing ones, and gain a deeper understanding of catalytic mechanisms. Machine learning has the potential to revolutionize the field of catalysis and pave the way for more sustainable and efficient chemical processes.
Machine learning has emerged as a powerful tool in catalysis research, enabling the discovery and optimization of catalysts with unprecedented efficiency. By leveraging large datasets, advanced algorithms, and computational modeling, researchers can accelerate the development of new catalysts, gain insights into catalytic mechanisms, and design more sustainable chemical processes. As the field continues to evolve, it is essential to address challenges related to data availability, model interpretability, and standardization.
With continued advancements in machine learning and catalysis research, we can expect to see further breakthroughs and innovations in the field, leading to a more sustainable and efficient future.
0 notes
Link
1 note
·
View note
Text
Animal Feed Industry - A Comprehensive Overview And A Vital Link In The Food Supply Chain
Global animal feed sector data book is a collection of market sizing information & forecasts, trade data, pricing intelligence, competitive benchmarking analyses, macro-environmental analyses, and regulatory & technological framework studies. Within the purview of the database, such information is systematically analyzed and provided in the form of outlook reports (1 detailed sectoral outlook report) and summary presentations on individual areas of research along with a statistics e-book.
Poultry Feed Market Analysis & Forecast
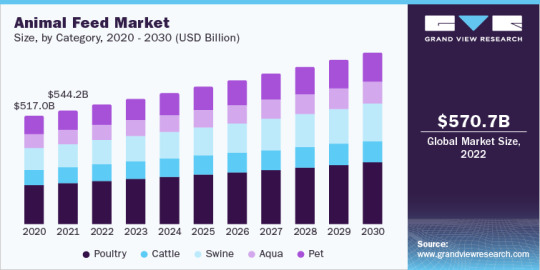
The global poultry feed market size was valued at 555.9 million tons in 2022, growing at a CAGR of 2.6% from 2023 to 2030. Poultry is considered as one of the most economical sources of protein, therefore, poultry products such as egg and meat are consistently witnessing growth in their demand. The market is likely to witness strong growth owing to increasing demand for protein-rich diet and increased production of feed across North America. The presence of well-established industry is another major driver triggering the product demand and, in turn, the overall market growth.
The market is fragmented and competitive with the presence of major global players. In the U.S., nearly 35 companies (federally inspected) are engaged in raising, marketing, and the processing of chicken. These companies are vertically integrated throughout the poultry value chain to ensure the quality of feed product. Brazil, the U.S., India, Mexico, Colombia, Thailand, and Russia registered the highest growth rate, in terms of chicken production, between 2014 and 2018. Demand for poultry products heavily depends upon the population and income factor. According to the Food and Agriculture Organization (FAO), the consumption of milk, eggs, and meat in low and middle-income economies increased by more than 300% in the last 30 years.
Access the Global Animal Feed Industry Data Book from 2023 to 2030, compiled with details by Grand View Research
Cattle Feed Market Analysis & Forecast
The global cattle feed market size was valued at 240.0 million tons in 2022 and is expected to grow at a compound annual growth rate (CAGR) of 2.9% from 2023 to 2030. Growing concerns of farmers regarding the yield and cattle health have resulted in high demand for high-quality ingredients and additives across the globe. According to the Veterinary Feed Directive (VFD), antibiotics in feed without a prescription from VFD should be avoided owing to infectious agents present in them.
Thus, as a replacement, ingredients like ionophores, prebiotics, probiotics, and enzymes are used as feed additives. Ionophores like Rumensin and Bovatec improve feed efficiency and reduce the risk of bloat and acidosis. XPC, Fermenten are some of the major products used to enhance productivity, minimize infections, and provide better immune to cattle. Thus, the cattle feed segment has tremendous growth opportunities due to a rise in the customization of meat and dairy products.
Swine Feed Market Analysis & Forecast
The global swine feed market size was estimated at 310.1 million tons in 2022 and is expected to expand at a CAGR of 3.3% over the forecast period. Increasing awareness among consumers concerning the quality of pork is compelling swine farmers to utilize additives that keep swine healthy as well as immune to diseases and disorders. Also, the use of enhanced additives is expected to reduce the occurrence of infections as well as illnesses in swine, thereby contributing to the increasing demand for the product worldwide.
Manufacturers in the market are constantly evaluating techniques to enhance growth performance as well as lower product costs. Physical forms, such as mash, crumbles, and pellets, play a crucial role in determining the quality and yield of pig meat. Rising urbanization and establishment of fast food restaurant chains are expected to augment the demand for pig meat. The trend is becoming increasingly prevalent in the emerging economies of Asia Pacific including China, India, and Malaysia. Population growth is another key factor that is expected to drive meat consumption globally. These factors together are expected to augment the demand for animal proteins, which, in turn, is likely to propel the demand for the product and additives over the forecast period.
Aquafeed Market Analysis & Forecast
The global aquafeed market size was valued at 40.7 million tons in 2022 and is anticipated to grow at a compound annual growth rate (CAGR) of 3.5% from 2023 to 2030. The growth is majorly driven by the rising consumption of aquafeed by species like carp, catfish, salmon, and shrimps, among others due to its rich protein content. The future of the global market is dependent on the increasing acceptance of aquafeed, which contains essential oils, feed acidifiers, natural extracts, and palatants, which are important for aquaculture species in their overall growth across each stage of development. The widespread fish mortality due to various infections, which are caused by parasites is driving the product demand. These feed products are consumed by various fish species, such as catfish, salmon, trout, tilapia, shrimps, largemouth bass, and eel.
These feeds for aquatic species are a source of omega-3 fatty acids and proteins. It improves the nutritional value of feed and offers several other benefits like improved growth rate, reduced mortality of various aquatic species, digestibility of proteins, and enhanced immune system. The COVID-19 pandemic had crippled the economies of the world and severely impacted the supply chain across key industries. The distribution of raw materials for aquafeed was affected by the pandemic due to which its production was hampered. In addition, strict lockdowns were imposed to curb the spread of the virus. Due to this, many manufacturing and production units were closed. Furthermore, there were rumors of transmission of the virus through poultry, which, in turn, hindered market growth.
Order Free Sample Copy of “Animal Feed Industry Data Book - Poultry Feed, Cattle Feed, Swine Feed, Aquafeed and Pet Food Market Size, Share, Trends Analysis, And Segment Forecasts, 2023 - 2030” published by Grand View Research
Pet Food Market Analysis & Forecast
The global pet food market size was valued at 31.8 million tons in 2022 and is anticipated to exhibit a compound annual growth rate (CAGR) of 3.6% from 2023 to 2030. The global pet food products have witnessed substantial demand over the past few years owing to the advent of online purchasing and notable contribution of e-commerce in shaping and strengthening the industry. Trends influencing the growth of this industry include launches of new products, online private brands, treats, and novel technologies.
Consumers do not prefer traditional products for their pet’s consumption. They try to comprehend the ingredients list and usually opt for healthier available alternative in the market. Consumers prefer made-to-order, frozen, and fresher meals for their pets. Although these types are comparatively expensive, customers are willing to pay more for healthy items for the consumption for their household pets. The outbreak of COVID-19, which has led to the closure of factories, production units, and manufacturing sites globally, there has been significant decline in the global demand for pet food ingredients and raw materials. Due to the ongoing pandemic, several production plants have been completely or partially shut down while others are running at reduced rates, which may result in a supply-demand gap globally.
Key players operating in the animal feed industry are –
• J.M. Smucker Company • The Hartz Mountain Corporation • Mars, Incorporated • Hill’s Pet Nutrition, Inc. • Nestlé Purina • LUPUS Alimentos • Total Alimentos • General Mills Inc. • WellPet LLC
#Animal Feed Industry Data Book#Poultry Feed Market Size#Cattle Feed Industry Trends#Swine Feed Market Size#Aquafeed Market Share#Pet Food Market Growth#Animal Feed Sector Report
0 notes
Text
Dolutegravir Prices | Pricing | Trend | News | Database | Chart | Forecast
Dolutegravir is an antiretroviral medication primarily used in the treatment of HIV/AIDS. As part of the class of drugs known as integrase inhibitors, Dolutegravir works by blocking the action of an enzyme called integrase, which the HIV virus uses to replicate. Since its approval by the U.S. Food and Drug Administration (FDA) in 2013, Dolutegravir has become a crucial component in many HIV treatment regimens. However, one of the most significant concerns around the world, especially in low-income and middle-income countries, has been the pricing of Dolutegravir. Pricing influences access to this essential medication, and efforts are ongoing to make it more affordable for those who need it most.
The cost of Dolutegravir varies widely depending on geographic location, patent laws, and whether the drug is branded or generic. In high-income countries such as the United States, Dolutegravir is often sold under the brand name Tivicay, manufactured by ViiV Healthcare. The brand-name version of Dolutegravir tends to be expensive, with prices in the U.S. being notably higher than in other regions. For example, a month's supply of Tivicay can cost thousands of dollars. This high cost is driven by research and development expenses, regulatory costs, and the need to recoup the investment in bringing a new drug to market. However, the high cost in wealthier nations often makes it difficult for those without adequate health insurance or government assistance to access the medication.
Get Real Time Prices for Dolutegravir : https://www.chemanalyst.com/Pricing-data/dolutegravir-1534
In contrast, many low- and middle-income countries rely on generic versions of Dolutegravir, which are significantly less expensive. Generic Dolutegravir became available after licensing agreements and patent waivers were negotiated between pharmaceutical companies, non-governmental organizations, and international agencies. These agreements allowed for the production of more affordable versions of the drug, making it accessible to larger populations. In countries such as India, which has a robust generic pharmaceutical industry, the price of Dolutegravir can be as low as a few dollars per month. This drastic reduction in price has been crucial in improving access to HIV treatment, particularly in regions that are heavily burdened by the epidemic, such as sub-Saharan Africa.
Global health organizations like the World Health Organization (WHO) and the Joint United Nations Programme on HIV/AIDS (UNAIDS) have played a pivotal role in advocating for lower prices for HIV medications, including Dolutegravir. The Medicines Patent Pool (MPP) is another initiative that has helped to lower the cost of Dolutegravir. By facilitating voluntary licensing agreements, the MPP enables generic drug manufacturers to produce and sell Dolutegravir at a fraction of the price of the branded version. These efforts have resulted in broader availability of the drug, but challenges remain in ensuring that everyone who needs Dolutegravir can afford it.
The introduction of Dolutegravir as part of first-line HIV treatment regimens in many countries has been a game-changer. Not only is Dolutegravir highly effective in suppressing the HIV virus, but it also has a high barrier to resistance, meaning that patients are less likely to develop resistance to the drug over time. Moreover, Dolutegravir has fewer side effects compared to older antiretroviral medications, making it a preferred option for many patients and healthcare providers. Given its effectiveness, there has been a global push to make Dolutegravir the standard of care in HIV treatment. However, pricing remains a barrier in some parts of the world, where even the generic versions may be out of reach for the most vulnerable populations.
Efforts to reduce the price of Dolutegravir are ongoing, with various international partnerships and coalitions working to negotiate lower costs. For example, the Global Fund to Fight AIDS, Tuberculosis and Malaria has been instrumental in securing lower prices for Dolutegravir through bulk purchasing agreements. These agreements allow for large quantities of the drug to be purchased at a reduced cost, which can then be distributed to countries in need. Additionally, some countries have implemented pricing controls or subsidies to make Dolutegravir more affordable for their populations. These strategies have been successful to varying degrees, depending on the political and economic landscape of each country.
Another factor influencing the price of Dolutegravir is the expiration of patents. As patents on the drug begin to expire in more countries, the opportunity for increased competition among generic manufacturers grows. This competition is expected to drive prices down even further, making Dolutegravir more accessible to a wider range of people. However, patent expirations are staggered across different regions, meaning that some countries may experience price reductions sooner than others. In countries where the patent is still in effect, advocacy efforts are focused on encouraging pharmaceutical companies to voluntarily lower their prices or enter into more licensing agreements that would allow for the production of generics.
In recent years, there has been a growing emphasis on the importance of affordable HIV treatment as part of global health initiatives. The United Nations has set ambitious goals for reducing the number of new HIV infections and ensuring that those living with HIV have access to treatment. Dolutegravir is central to these efforts, but its price remains a critical issue. Without continued pressure on pharmaceutical companies and governments, there is a risk that some populations will continue to be left behind in the fight against HIV/AIDS.
In conclusion, the pricing of Dolutegravir reflects a complex interplay of factors, including patent laws, production costs, and global health policies. While progress has been made in making the drug more affordable, particularly in low- and middle-income countries, there is still work to be done to ensure that everyone who needs Dolutegravir can access it. The future of HIV treatment depends not only on the development of new and effective medications but also on the ability to make these treatments affordable and accessible to all. Through continued international cooperation and advocacy, it is possible to further reduce the price of Dolutegravir and ensure that it reaches those who need it most.
Get Real Time Prices for Dolutegravir : https://www.chemanalyst.com/Pricing-data/dolutegravir-1534
Contact Us:
ChemAnalyst
GmbH - S-01, 2.floor, Subbelrather Straße,
15a Cologne, 50823, Germany
Call: +49-221-6505-8833
Email: [email protected]
Website: https://www.chemanalyst.com
#Dolutegravir#Dolutegravir Price#Dolutegravir Prices#Dolutegravir News#Dolutegravir Market#Dolutegravir Pricing
0 notes
Text
Silage Additives Market Share Growing High CAGR During 2023-29
The Silage Additives Market Report provides an exhaustive analysis of the growth drivers, current trends, restraining forces, and opportunities present in the market.
The global Silage additives market size was valued at USD 2,523.2 million in 2022 and is poised to grow at a significant CAGR of 4.6% during the forecast period 2023-29. It also includes market size and projection estimations for each of the five major regions from 2023 to 2029. The research report includes historical data, trending features, and market growth estimates for the future. Furthermore, the study includes a global and regional estimation and further split by nations and categories within each region. The research also includes factors and barriers to the Silage additives market growth, as well as their impact on the market's future growth. The report gives a comprehensive overview of both primary and secondary data.
View the detailed report description here - https://www.precisionbusinessinsights.com/market-reports/silage-additives-market
The global Silage additives market segmentation:
1) By Product Type : Organic acids, Enzymes, Sugars, Inoculants, Others.
2) By Silage Crop : Alfalfa, Oats, Rye, Barley, Corn, Sorghum, Others.
3) By Formulation : Dry, Liquid.
4) By Application : Inhibition, Stimulation, Moisture Absorption, Others.
The primary factors of the Silage additives market drivers are the increasing focus of governments on feed cost reduction, and boosting animal productivity. The Silage additives market report helps to provide the best results for business enhancement and business growth. It further helps to obtain the reactions of consumers to a novel product or service. It becomes possible for business players to take action for changing perceptions. It uncovers and identifies potential issues of the customers. It becomes easy to obtain the reactions of the customers to a novel product or service. It also enlightens further advancement, so it suits its intended market.
The Silage additives marketresearchreport gives a comprehensive outlook across the region with special emphasis on key regions such as North America, Europe, Asia Pacific, Latin America, and the Middle East and Africa. North America was the largest region in the Silage additives market report, accounting for the highest share in 2022. It was followed by Asia Pacific, and then the other regions. Request sample report at - https://www.precisionbusinessinsights.com/request-sample/?product_id=21129
The important profiles and strategies adopted by Silage additives market key players are BASF SE (Germany) DuPont, Pioneer (DuPont) (U.S.) Lallemand, Inc. (U.S.) Schaumann BioEnergyGmbH (Germany) Hansen A/S (Denmark) Biomin (Austria) Volac International Limited (UK) Micron Bio-Systems (U.S.) American Farm Products (U.S.) Nutreco N.V. (Netherlands) BioZyme, Inc. (U.S.). covered here to help them in strengthening their place in the market.
About Precision Business Insights: We are a market research company that strives to provide the highest quality market research insights. Our diverse market research experts are enthusiastic about market research and therefore produce high-quality research reports. We have over 500 clients with whom we have a good business partnership and capacity to provide in-depth research analysis for more than 30 countries. In addition to deliver more than 150 custom solutions, we already have accounts with the top five medical device manufacturers.
Precision Business Insights offers a variety of cost-effective and customized research services to meet research requirements. We are a leading research service provider because of our extensive database built by our experts and the services we provide.
Contact:
Mr. Satya
Precision Business Insights | Toll Free: +1 866 598 1553
Email: [email protected] Kemp House, 152 – 160 City Road, London EC1V 2NX Web: https://precisionbusinessinsights.com/ | D U N S® Number: 852781747
#silage additives market size#silage additives market share#silage additives market growth#silage additives market analysis#silage additives market trends
0 notes
Text
Molecular Enzymology and Drug Targets
Molecular Enzymology and Drug Targets journal aims is to publish the work of designing and synthesis of enzymes and high unmet medical need are based on innovative drug targets. This is an international journal for rapid dissemination of significant data related to enzymes and their use in biotechnology and drug discovery.
The journal is an interdisciplinary medium serving several branches of life sciences. This Journals publishes original articles, short communications, notes, and mini reviews. Full length reviews are only published after invitation from the editorial board. Preliminary studies are inappropriate for publishing in Journal of Molecular enzymology and Drug Targets, unless the authors report the significance of their findings, in that case, the manuscript can be accepted. Average first decision of submitted manuscript is expected to be 14 days.
Submit manuscript as an e-mail attachment to the Editorial Office at [email protected]

0 notes