
Don't wanna be here? Send us removal request.
Statistics
We looked inside some of the posts by alsdeanna0209 and here's what we found interesting.
Average Info
Notes Per Post
5
Likes Per Post
5
Reblog Per Post
0
Reply Per Post
0
Time Between Posts
13 days ago
Number of Posts By Type
Text
6
Link
2
Last Seen Tumblr Blogs





Fun Fact
In 2020, 27% of US Tumblr users had an annual household income of over $100,000.
Text
The Typical Course of Treatment for ALS
Amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig’s disease, is a progressive neurodegenerative disease characterized by muscle weakness and atrophy, which can affect breathing, swallowing, and talking. The most common cause of death among people with ALS is respiratory failure due to the muscles involved in breathing becoming weak. While there’s no cure for ALS and treatment options are limited, some therapies exist to mitigate the symptoms of ALS and increase life expectancy and quality of life in patients with the condition. Here’s an overview of what you can expect from the course of treatment for ALS.
While there is no cure for amyotrophic lateral sclerosis (ALS), treatments may help slow its progression. People with ALS usually are treated by a team that includes at least one neurologist, one physician who specializes in treating muscle-related disorders (neuromuscular medicine specialist), and one physical therapist. Other specialists involved in treatment include pulmonologists, gastroenterologists, ophthalmologists, otolaryngologists, and orthopedic surgeons. Supportive care also plays an important role in ALS treatment.
Examples of supportive care include:
Respiratory support . Some people with ALS need to use ventilators to breathe and/or supplemental oxygen. In some cases, individuals also receive noninvasive positive pressure ventilation through masks or mouthpieces or tracheostomy tubes that provide continuous breathing assistance.
Health care team . People with ALS may benefit from physical therapy, speech therapy, occupational therapy, and nutritional counseling. A dietitian can help create an individualized meal plan based on a person’s needs.
Medications . There are no medications currently available to treat ALS itself. However, medications are available to manage symptoms like spasticity, depression and other psychological issues, drooling and excessive saliva production (dysphagia), constipation and other bowel problems, difficulty swallowing (dysphagia), excessive urination (polyuria), difficulty speaking (dysarthria), dizziness, lack of coordination or tremors.
Other therapies. Treatments may include acupuncture and supportive equipment such as splints, braces, crutches, walkers or wheelchairs.
Rehabilitation . Physiatrists, also known as rehabilitation physicians, often are involved in providing physical and occupational therapy.
Occupational therapists work with people with ALS to maintain function, such as independent feeding and self-care skills.
Physical therapists may focus on strength training or on range-of-motion exercises that keep muscles flexible and decrease pain.
Studies have shown that some individuals can increase their daily walking ability by using continuous positive airway pressure (CPAP) devices. This treatment involves blowing pressurized air into a mask placed over an individual’s mouth and nose while he or she sleeps at night. The goal is to reduce respiratory disturbances during sleep that can worsen breathing problems throughout the day. If you need help getting around, an occupational therapist can recommend adaptive equipment such as wheelchairs, walkers, bathroom equipment and seating systems for wheelchairs and cars.
An exercise physiologist can design an aerobic workout program that may slow down muscle loss caused by ALS.
A speech pathologist can create exercises to slow language deterioration. And although there currently are no drugs available specifically to treat speech problems associated with ALS, several medications commonly used for other disorders—such as antidepressants, antipsychotics and anticonvulsants—may be helpful for treating symptoms related to motor neuron disease. This includes improvement in slurred speech and dysarthria (speech disorder).
The course of amyotrophic lateral sclerosis (ALS) depends on a number of factors, including which areas of your body are affected by muscle weakness.
0 notes
Text
What exactly is ALS?
Cause of ALS

ALS causes the death of motor neurons, which are responsible for controlling voluntary movements and muscle control. A voluntary movement is defined as one in which the muscles of the arms and legs are directed by the mind rather than by the body. Lower motor neurons transmit information from the spinal cord to the muscles throughout the body, whereas upper motor neurons transmit information from the brain to the spinal cord. Motor neurons are found in both the spinal cord and also the brain. ALS affects the upper and lower motor neurons in the brain, spinal cord, and brain stem, which are responsible for movement. Muscle tension is caused by degeneration of the upper motor neuron, which leads in spasticity, whereas muscle weakness, atrophy (muscle shrinkage), and twitching are caused by degeneration of the lower motor neuron, which results in spasticity. The slow degeneration of motor neurons in the brain and spinal cord causes the neurons to atrophy over time, finally resulting in the neurons being killed. When motor neurons are damaged, the brain is no longer capable of initiating, moving, or controlling muscle activity. As a result, ALS patients' muscles get smaller and weaker within a few years of being diagnosed, making it difficult for them to speak, eat, move upstairs, reach for objects, and carry out their daily activities effectively.
Prognosis for ALS

Several variables have been recognized as being responsible for the wide range of ALS symptoms. For example, as compared to individuals with spinal beginning for ALS, which affects the limbs and trunk of patients, those with early bulbar symptoms had the lowest survival prognosis. However, the prognosis for respiratory onset ALS which affects the respiratory system first is extremely bleak. The patient's prognosis is made worse by the type of ALS he or she has. Another factor that makes the prognosis worse is the onset of the disease later in life. Those who have it before they reach their forties live substantially longer. The kind of ALS that is whether hereditary or not, has an impact on the patient's prognosis. Patients with a hereditary form of ALS caused by the gene mutation A4V, which affects the SOD1 enzyme, have a 12-month survival rate and other mutations in the same gene result in an illness that is less aggressive.
Weakness in the neck muscle might indicate rapid decline in a person's ability to do ordinary chores and a shorter life. Experts in the area of ALS disagree on whether three other factors influence the disease's course and patient survival. Gender, time needed to diagnose a person having symptoms, and whether ALS manifests itself in the upper or lower limbs are all factors to consider. The question of whether a delay in diagnosis affects the course of ALS and the survival rate of patients is complicated by the possibility that individuals who see a doctor later may have a condition that progresses more slowly. Aside from the movement issues that come with ALS, there has been growing acknowledgment that the disease also causes movement-related issues like thinking. Around 14% of ALS patients acquire frontotemporal dementia, meaning a lower life expectancy as a result of the disease, while up to 50% exhibit mild cognitive impairment.
0 notes
Text
Treatments & Medications for ALS

Lou Gehrig's illness is the most common form of ALS (amyotrophic lateral sclerosis). He was a big league baseball player with the New York Yankees and one of the best sluggers of all time, hitting 493 home runs before being forced to retire in 1939 due to ALS. Gehrig revealed to a gathering of 62,000 fans on July 4, 1939, that he was retiring from baseball due to the sickness. He died on June 2, 1941, less than two years later. He was 36 years old at the time.
At the time, most people had never heard of Lou Gehrig's disease, and it was inextricably tied to Gehrig's death. Lou Gehrig is not the only well-known individual who has been diagnosed with Lou Gehrig's disease (ALS). Senator Jacob Javits and Stephen Hawking, the world-renowned scientist, Jim "Catfish" Hunter, a former professional baseball player, and other well-known figures have all suffered from Lou Gehrig's disease (ALS).
As many as 30,000 people are affected by Lou Gehrig's disease (ALS) in the United States today, with 5,000 new cases identified each year. Despite the fact that it can manifest itself at any age, the vast majority of cases are diagnosed in people between the ages of 40 and 70 years old. A higher proportion of people over the age of 60 are affected by this condition than younger people.
Treatment options for ALS

There is no cure for ALS, and no effective therapies exist to slow or stop the disease's development. The major symptoms of ALS are treated with two medications: Tiglutik and Rilutek (riluzole). Muscle cramps, excessive saliva and phlegm, uncontrollable weeping or laughing, discomfort, depression, sleep problems, and constipation may all be treated with other therapies and medicines.
Other treatments, in addition to medicines, may be beneficial:
Physical therapy can help people gain independence and feel more secure. Low-impact exercise like walking, swimming, and stationary bicycling helps to build muscles, enhance cardiovascular health, and combat weariness and depression. Physical activity also increases range of motion, avoids stiffness, and reduces muscular shortening.
Practical difficulties such as ramps, braces, walkers, and wheelchairs may benefit from occupational therapy. Occupational therapists can also aid with additional assistive gadgets and techniques to better handle everyday chores like clothing and hygiene.
Communication problems can occasionally be helped by speech therapy. Therapists may also educate caregivers how to interact with their loved ones using computer-based speech synthesizers.
When chewing and swallowing become difficult, nutritional supplementation may be beneficial. Nutritional therapists can assist caregivers in planning and preparing meals that offer calories, fiber, and water while reducing choking risks.
Some patients may require ventilators to inflate and deflate their lungs as the disease progresses.
ALS research, including clinical trials, is underway in the hopes of finding more effective therapies. Here are some websites to help you locate a clinical study in your area:
The ALS Association and the Northeast ALS Consortium are two organizations that work together to help people with ALS.
Clinicaltrials.gov
Center Surveillance
The Centers for Disease Control and Prevention (CDC)
Medication for ALS
The United States Food and Drug Administration (FDA) has authorized two medicines for the treatment of ALS.
youtube
0 notes
Text
Amyotrophic Lateral Sclerosis
Both symptoms-relieving therapy and disease-modifying are available, as well as new therapeutic options.
Amyotrophic lateral sclerosis (ALS) is a terminal neurological illness that begins with weakness in the limbs and progresses to dysarthria, dysphagia, and dyspnea. Pseudobulbar affect, sialorrhea, tiredness, spasticity, cramping, and weakness are just a few of the distressing symptoms. Death usually occurs 2 to 5 years after the beginning of symptoms, and is caused by a decrease in swallowing and breathing. The loss of central and peripheral motor neurons causes symptoms. The actual pathophysiology of ALS is still being researched, and the illness is complicated, with numerous theories offered. There is no cure; however, the Food and Drug Administration (FDA) has authorized two disease-modifying therapies (DMTs) that reduce disease development but do not enhance strength or function. A variety of medicines are also used to treat particular symptoms, frequently without an FDA-approved indication (i.e. “off label”). Clinical studies are underway for a number of potential therapeutic medicines, some of which have shown encouraging results. This article explores developing therapy options and reviews existing medicines to halt disease development and lessen symptom severity.
Pathophysiology

The aggregation of proteins, particularly the TAR DNA binding protein 43, is a plausible explanation for the genesis and development of the ALS disease process (TDBP43). Another prominent theory argues that aberrant increases in reactive oxygen species arise, causing the cells to be injured. 4 Excess glutamate-induced excitotoxicity, immune-mediated inflammatory responses, and mitochondrial dysfunction linked to superoxide dismutase 1 (SOD1) mutations are among the other possibilities. These ideas are not mutually exclusive, and various variables, possibly both genetic and environmental, are likely to have a role.
Approved Disease-modifying Treatment
Riluzole. Riluzole, a glutamate inhibitor, has been demonstrated to have a neuroprotective effect, most likely by reducing glutamate transmission and inhibiting harmful excess glutamate. Riluzole was authorized after a randomized clinical trial revealed that riluzole had a considerably better 1-year survival rate (74%) than placebo (58 percent). 5 Additional trials verified the efficacy of riluzole in increasing 1-year survival rates and determined that 100 mg daily in two 50-mg doses is the optimum therapeutic dosage. 1 Fatigue, dizziness, and gastrointestinal problems are the most frequent side effects (eg. nausea, abdominal pain, or diarrhea). These effects may disappear if the dosage is reduced to 50 mg once a day. Riluzole can cause increased liver enzymes, which usually return to normal after the drug is stopped, and in rare circumstances, neutropenia.

Edaravone. Approved in 2017, edaravone is expected to alleviate ALS development by neuroprotective removal of free radicals. 4 Although it should be noted that this benefit occurred specifically in participants who were in the early stages of ALS, and edaravone efficiency will decrease as the ALS progresses, edaravone treatment resulted in significantly lower declines on the revised ALS functional rating scale (ALSFRS-R) compared to placebo in clinical trials. 4 Edaravone is given intravenously in the regular doses of 60 mg spread out over an hour. The treatment starts with 14 days of daily infusions, followed by 14 days without treatment, and then 10 days of infusions every month (which may be split into 5 days, 2 days off, and then another 5 days). Bruising at the injection site, headaches, and gait abnormalities are all possible side effects. Thrombosis, bleeding, and infection are among the possible complications with intravenous administration systems (such as port-a-caths and central lines). Most insurance providers in the United States have a stringent set of reimbursement requirements for edaravone medication that closely resembles the clinical trials enrollment criteria.
youtube
0 notes
Text
ALS: Types and Diagnosis

ALS symptoms are unclear. Early symptoms may be misdiagnosed as cold or flu, tiredness, sadness, or other illnesses. Testing for ALS usually delayed until other illnesses are ruled out.
The first step in diagnosing ALS is a physical examination and history. The examiner will search for symptoms of MND. A general physical exam, a neurological exam that may involve a manual muscle test to examine every area of your body's function and feeling, and a physical evaluation of your cranial nerves will be included.
You may get EMG and nerve conduction tests (NCS). EMG measures your muscles' capacity to move. NCS tests your nerves' health. These tests will likely be run concurrently. An MRI of the brain and spinal cord is conducted to examine your body's physiology and rule out any other abnormalities causing your symptoms. An ALS patient's MRI is usually normal. Blood and urine tests will be done to rule out other conditions.
Some illnesses have symptoms with ALS. These can be:
Fasciculations and the muscle cramps occur also in benign conditions Polio and PPS
Human immunodeficiency virus (HIV)
HTLV or Human T-cell leukemia viruses
Multiple Sclerosis
Multifocal motor neuropathy
Polio and post-polio syndrome
Spinal and the bulbar muscular atrophy
West Nile Virus
The Functional Rating Scale-Revise of ALS is used to assess an individual's ALS-related function (ALSFRS-R). This scale assesses changes in condition as time passes by. This includes: (1) speaking; (2) salivation; (3) swallowing; (5) cutting food and using utensils (with or without gastrostomy); (6) clothing and hygiene; (7) turning in bed and changing bedclothes; (10) breathing. Total rating ranges from 0 to 40. The lower the total score, the more ALS. This scale should be used for all assessments to ensure uniformity.
ALS Types

There are several different types of ALS, with many of them being connected to a genetic defect. These subgroups have an impact on the symptoms and development of the condition. Steven Hawking, a scientist, has lived 55 years after being diagnosed with Lou Gehrig's disease (ALS). ALS treatment and research seldom differentiate between subtypes. Individualized ALS care is available for patients suffering from any subtype of the disease.
There are two ALS development patterns.
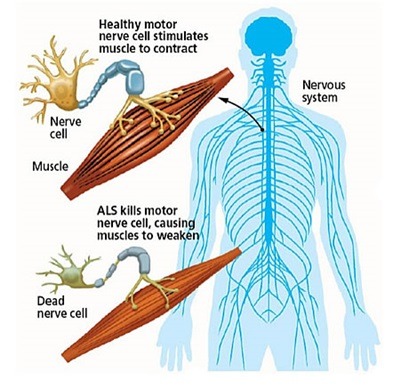
ALS is most commonly sporadic. In some situations, ALS develops without cause. There is no recognized cause or familial risk. It is rare to inherit or pass on the illness to offspring. 90% of ALS cases are sporadic.
A defect in any of 12 identified genes causes familial ALS. C9orf72, SOD1, TARDBP, and FUS are often linked. The gene C9ORF72 (chromosome 9 open reading frame 72) is linked to ALS and frontal lobe dementia. People with this gene problem can have both illnesses.
Inheritance of ALS from a parent can affect biological offspring. 5-10% of instances have genetic irregularities, while spontaneous ALS has no known inheritance.
There are a variety of theories on how ALS develops. Toxin exposure, strenuous exercise, and immunological dysfunction are all examples of such conditions. Neurodegeneration that is not caused by inflammation is another option. Destruction of myelin sheaths is another potential (loss of myelin coating covering the nerve). However, none of these hypotheses are convincing in the case of ALS.
0 notes
Text
ALS Disease: What is it and how do I find out more?

ALS disease, or amyotrophic lateral sclerosis, is a neurological disease affecting the brain and spinal cord. People with ALS disease will experience motor neuron degeneration over time, causing decreased movement control and muscle mass, as well as problems with coordination and speaking abilities. There’s no known cure or ALS treatment that can reverse the effects of ALS disease, but there are programs to help control pain, improve quality of life, and support those suffering from the effects of this debilitating disease. In order to find out more about ALS disease, keep reading to learn about what it is and what you can do to find out more.
1) Symptoms

Symptoms of ALS typically develop gradually, starting with muscle twitching or weakness in one limb. Eventually, these symptoms spread to other parts of your body, such as your hands or feet. Many people who have ALS report muscle cramps and pain in their arms, legs, chest and back. Affected individuals may also experience muscle spasms that cause twisting movements of your body (spasticity). About one-third of people with ALS eventually become totally paralyzed, while others retain some ability to move even after their diagnosis.
2) Causes

ALS disease (Amyotrophic Lateral Sclerosis) is a progressive neurodegenerative disease that affects nerve cells in your brain and spinal cord. The nerve cells are responsible for controlling muscle movement, which means you may experience weak muscles, muscular atrophy, or paralysis if you suffer from ALS.
3) Common Treatments

Treatments for ALS are often centered around managing pain or keeping patients safe, since there are no cures for these diseases. For instance, people with ALS may need to use special equipment to help them breathe or eat. A feeding tube can ensure they’re getting adequate nutrition if their swallowing muscles don’t work properly. Pain medications can relieve other types of discomfort associated with ALS, such as joint pain or muscle stiffness.
4) Understanding ALS

ALS, short for amyotrophic lateral sclerosis, also known as Lou Gehrig’s disease, is a neurodegenerative disease that attacks nerve cells in your brain and spinal cord. It causes you to lose control of voluntary muscle movement. With certain types of ALS, even basic functions like breathing can become difficult or impossible. At least 12 times more common among men than women, ALS typically starts when you’re between 40 and 70 years old; there’s no way to prevent or cure it.
Final Thoughts
Although only 5% of Americans have heard of ALS, also known as Lou Gehrig’s disease, each year in America, about 12,000 new cases are diagnosed. Those diagnosed with ALS lose motor skills over time. Some die within 3 years of diagnosis; others survive for 30 years or longer. The average life expectancy after diagnosis is two to five years. There are no medical treatments that stop or reverse ALS symptoms.
youtube
1 note
·
View note
Link
1 note
·
View note
Link
3 notes
·
View notes